Useful to compare taxonomy from two different source/db/algo
Usage
rainplot_taxo_na(
physeq,
ranks = NULL,
min_nb_seq = 0,
merge_sample_by = NULL,
sample_colored = FALSE,
sample_linked = FALSE,
...
)Arguments
- physeq
(required) A
phyloseq-classobject obtained using thephyloseqpackage.- ranks
(character or integer vector, default NULL) The ranks to include in the rainplot. If left to NULL, all ranks are used. Each rank can be defined either by integer for the index or by its full names (exactly matching the colnames of the
tax_tableslot).- min_nb_seq
(integer, default 0) Minimum number of sequences to filter out taxa with low abundance.
- merge_sample_by
(character, default NULL) A vector to determine which samples to merge using
merge_samples2()function. Need to be inphyseq@sam_data.- sample_colored
(logical, default FALSE) If TRUE, points are colored by samples.
- sample_linked
(logical, default FALSE) If TRUE, points are linked by samples.
- ...
Additional arguments passed to
ggrain::geom_rain().
Examples
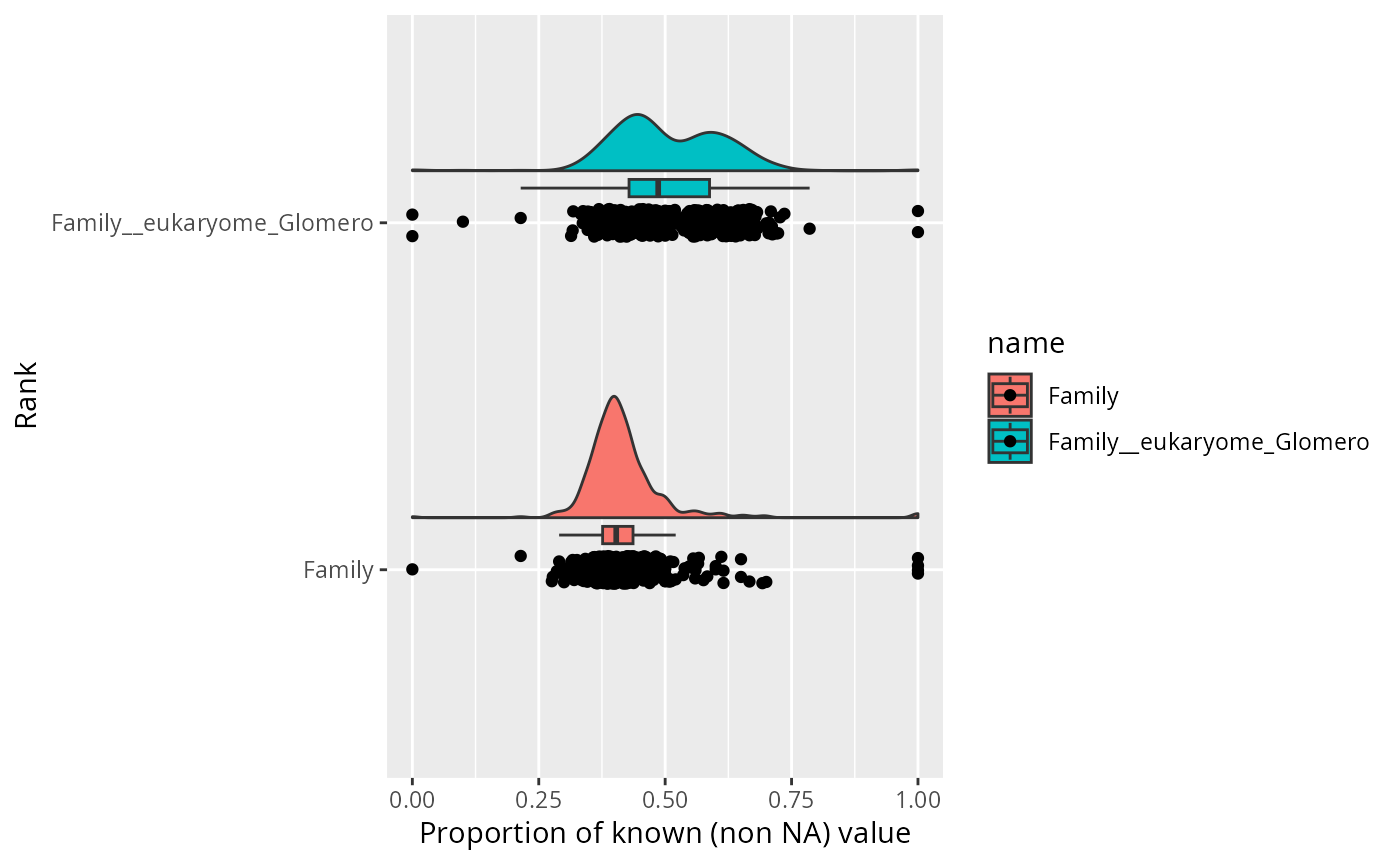
rainplot_taxo_na(subset_taxa_pq(Glom_otu, taxa_sums(Glom_otu) > 5000),
ranks = c("Family", "Family__eukaryome_Glomero")
)
#> Cleaning suppress 0 taxa ( ) and 1 sample(s) ( samp_Blanc-PCR-racines ).
#> Number of non-matching ASV 0
#> Number of matching ASV 1147
#> Number of filtered-out ASV 955
#> Number of kept ASV 192
#> Number of kept samples 443
#> Registered S3 methods overwritten by 'ggpp':
#> method from
#> heightDetails.titleGrob ggplot2
#> widthDetails.titleGrob ggplot2
 if (FALSE) { # \dontrun{
rainplot_taxo_na(Glom_otu)
Glom_otu@sam_data$tmt_type <- paste0(Glom_otu@sam_data$Tmt, "_", Glom_otu@sam_data$Type)
rainplot_taxo_na(
Glom_otu,
merge_sample_by = "tmt_type",
sample_colored = TRUE,
sample_linked = TRUE
)
rainplot_taxo_na(Glom_otu, ranks = c(4, 12), rain.side = "f1x1")
rainplot_taxo_na(
Glom_otu,
ranks = c(6, 14),
rain.side = "f1x1",
sample_linked = TRUE
) +
theme(legend.position = "none")
} # }
if (FALSE) { # \dontrun{
rainplot_taxo_na(Glom_otu)
Glom_otu@sam_data$tmt_type <- paste0(Glom_otu@sam_data$Tmt, "_", Glom_otu@sam_data$Type)
rainplot_taxo_na(
Glom_otu,
merge_sample_by = "tmt_type",
sample_colored = TRUE,
sample_linked = TRUE
)
rainplot_taxo_na(Glom_otu, ranks = c(4, 12), rain.side = "f1x1")
rainplot_taxo_na(
Glom_otu,
ranks = c(6, 14),
rain.side = "f1x1",
sample_linked = TRUE
) +
theme(legend.position = "none")
} # }