Draws a Venn diagram showing shared and unique taxa (or higher-rank groups) across 2 to 4 levels of a sample variable, using only ggplot2 (no external Venn diagram package needed).
When taxonomic_rank is a character vector of length > 1 (the
default), all ranks are displayed in a single combined figure using
the patchwork package (must be installed). Set combine = FALSE to
get a named list of individual plots instead.
For 2 and 3 groups, circles are used. For 4 groups, ellipses are used to ensure all intersection regions are representable.
Usage
simple_venn_pq(
physeq,
fact = NULL,
min_nb_seq = 0,
taxonomic_rank = c("Class", "Order", "Family", "Genus", "Species"),
na_remove = TRUE,
count_type = c("rank_taxa", "rank", "taxa", "sequences"),
add_nb_samples = TRUE,
fill_alpha = 0.3,
border_size = 0.8,
text_size = 4,
scale_text = FALSE,
hide_zero = TRUE,
label_size = 4.5,
colors = NULL,
labels = NULL,
show_na_count = FALSE,
count_taxa = TRUE,
match_by = c("refseq", "names"),
combine = TRUE,
verbose = TRUE
)Arguments
- physeq
(phyloseq or list_phyloseq, required) A phyloseq object, or a list_phyloseq object. When a list_phyloseq is provided, it is first merged into a single phyloseq using
merge_lpq()(each original phyloseq becomes one sample) and thefactparameter is automatically set to"source_name".- fact
(character, required when
physeqis a phyloseq) Name of a variable insample_data(physeq)defining the groups (2-4 levels). Ignored whenphyseqis a list_phyloseq.- min_nb_seq
(integer, default 0) Minimum total read count for a taxon to be considered present in a group. A taxon must have strictly more than
min_nb_seqreads in a group to be included.- taxonomic_rank
(character or NULL) Taxonomic rank(s) at which to aggregate (via
phyloseq::tax_glom()) before computing the Venn diagram. Defaults to all standard ranks (Kingdom through Species). UseNULLto skip aggregation and work at ASV/OTU level.- na_remove
(logical, default TRUE) Remove samples with NA in
factand, when aggregating, taxa with NA attaxonomic_rank.- count_type
(character, default
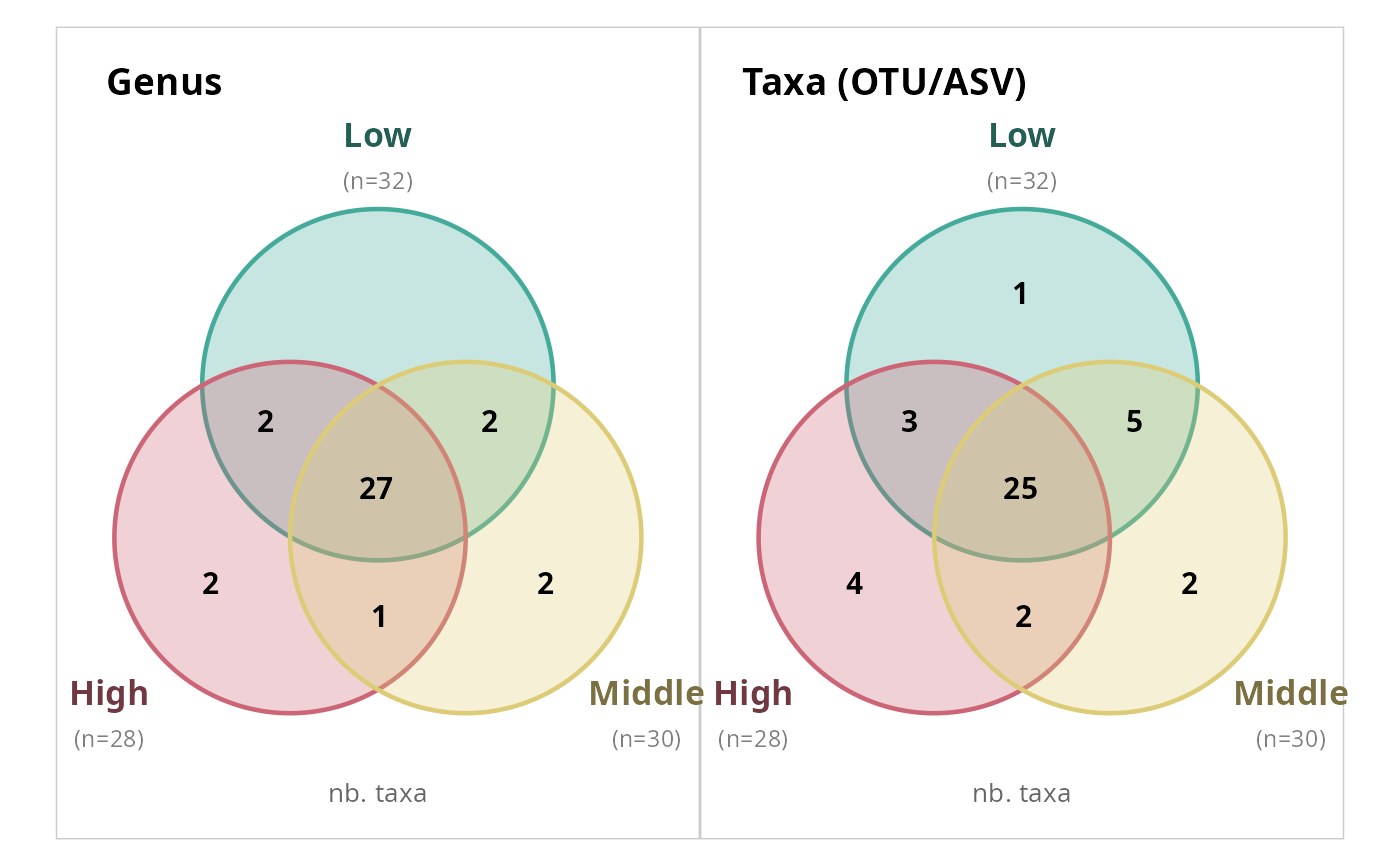
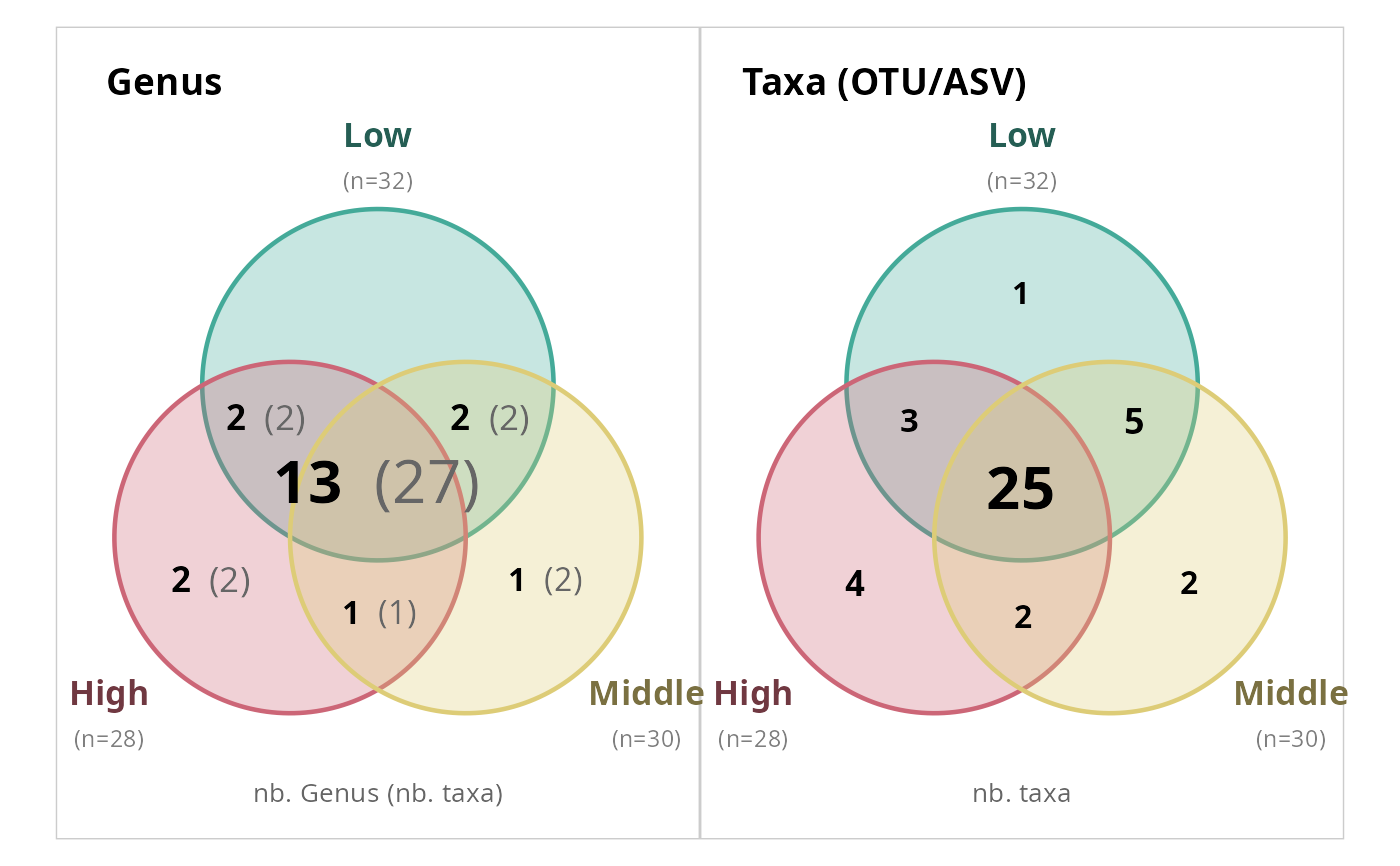
"rank_taxa") What to count in each Venn region. One of:"rank": number of unique taxonomic levels (e.g. number of shared Classes). This is the default."taxa": number of ASVs/OTUs assigned to the shared taxonomic levels."sequences": total number of reads for ASVs/OTUs assigned to the shared taxonomic levels."rank_taxa": shows both rank and taxa counts as"nb_rank (nb_taxa)". Ignored whentaxonomic_rankisNULL(ASV-level), where"rank"and"taxa"are equivalent.
- add_nb_samples
(logical, default TRUE) Append sample count to group labels.
- fill_alpha
(numeric, default 0.3) Fill transparency for shapes.
- border_size
(numeric, default 0.8) Border line width.
- text_size
(numeric, default 4) Base size of count labels inside regions.
- scale_text
(logical, default FALSE) If
TRUE, scale the size of count labels proportionally to the count value. Thetext_sizeparameter then acts as the base (minimum) size.- hide_zero
(logical, default TRUE) If
TRUE, hide count labels that are zero (or"0 (0)"whencount_type = "rank_taxa").- label_size
(numeric, default 4.5) Size of group name labels.
- colors
(character or NULL) Vector of colors, one per group. Defaults to a 4-color qualitative palette.
- labels
(character or NULL, default NULL) Custom labels for the groups, in the same order as the levels of
fact(or the list_phyloseq names). Must have the same length as the number of groups. WhenNULL, the original level names are used. Not that the order is the one of the levels infact.- show_na_count
(logical, default FALSE) If
TRUE, display the number of taxa withNAat the chosentaxonomic_rankin the bottom-left corner of the plot. Whencount_type = "taxa", the sum of all Venn region counts plus the NA count equalsntaxa(physeq). Ignored whentaxonomic_rankisNULL.- count_taxa
(logical, default TRUE) If
TRUE, append a"Taxa"panel to the Venn diagram showing shared and unique individual taxa (ASVs/OTUs) alongside the aggregated taxonomic ranks. A temporaryTaxacolumn is added to the tax_table with each taxon's name as its value. Ignored whentaxonomic_rankisNULL.- match_by
(character, default
"refseq") Passed tomerge_lpq()whenphyseqis a list_phyloseq. One of"refseq"or"names".- combine
(logical, default TRUE) When
taxonomic_rankhas length > 1, combine plots into a single patchwork figure. Set toFALSEto return a named list of individual ggplot objects. Requires the patchwork package.- verbose
(logical, default TRUE) Print a message when no taxa meet the criteria.
Value
A ggplot2 object (single rank), a patchwork object (multiple
ranks with combine = TRUE), or a named list of ggplot2 objects
(multiple ranks with combine = FALSE).
Examples
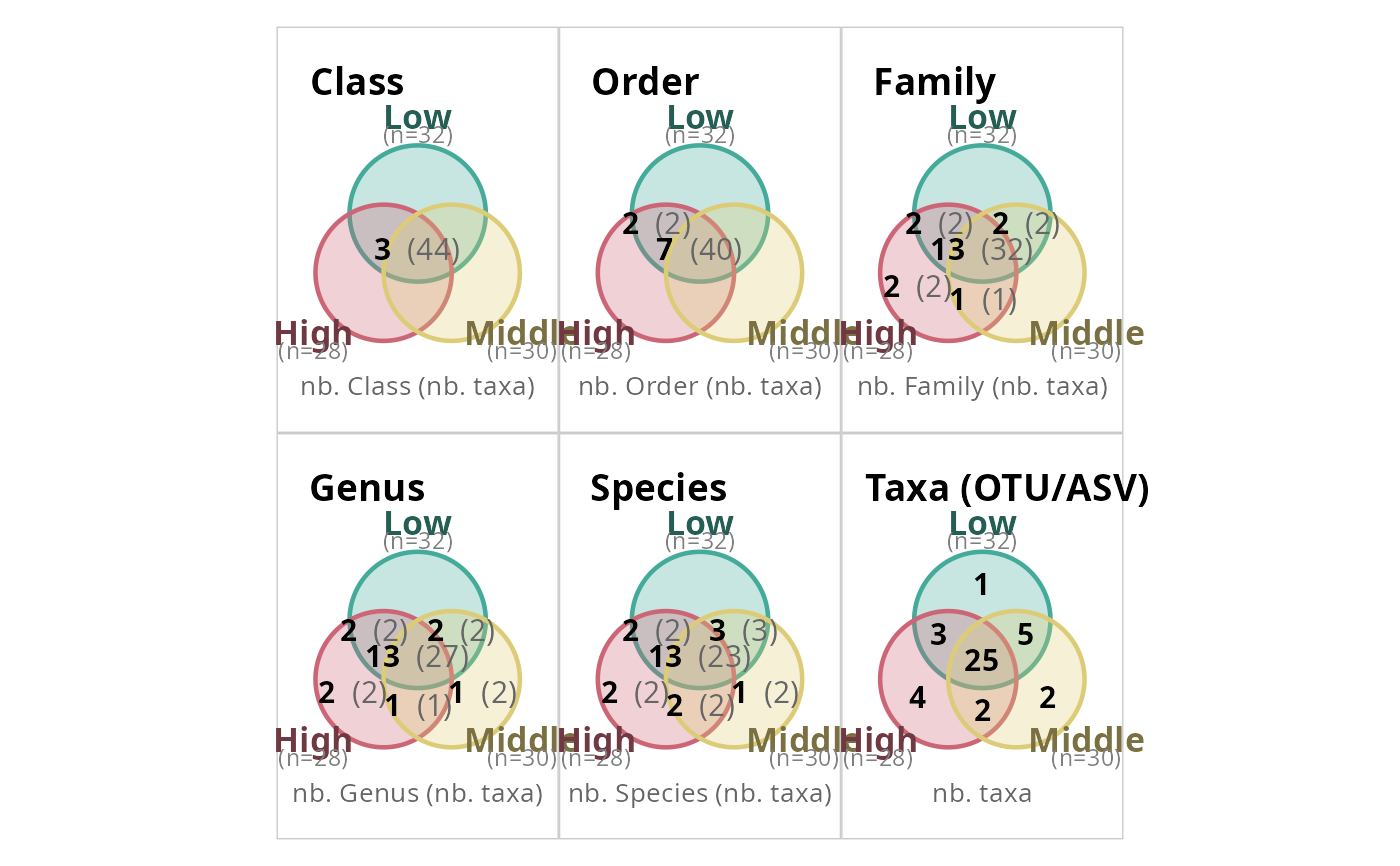
# Default: all ranks combined in one figure
simple_venn_pq(data_fungi_mini, "Height")
 # At genus level only
simple_venn_pq(data_fungi_mini, "Height", taxonomic_rank = "Genus")
# At genus level only
simple_venn_pq(data_fungi_mini, "Height", taxonomic_rank = "Genus")
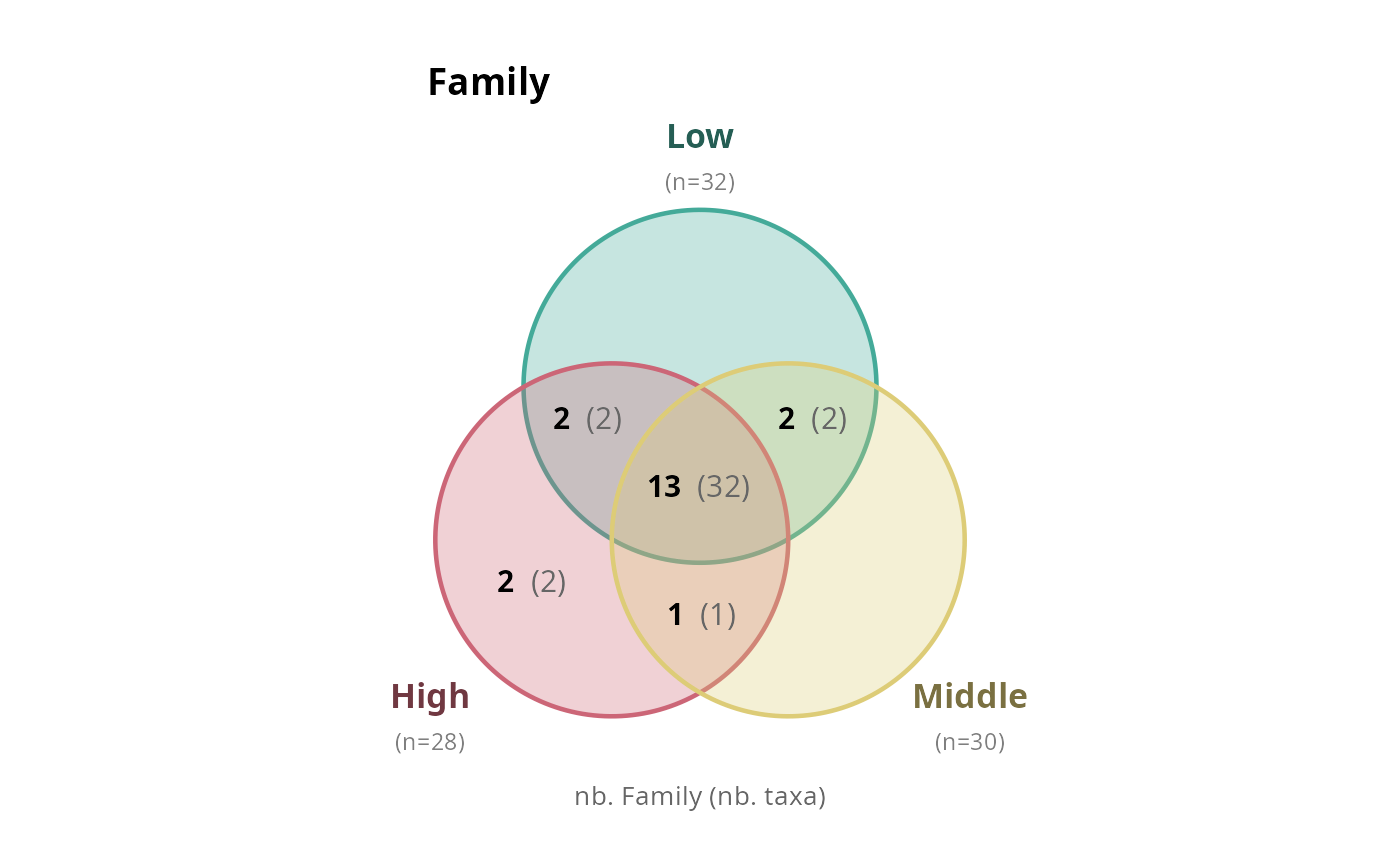
 # Multiple ranks as a list
plots <- simple_venn_pq(
data_fungi_mini, "Height",
taxonomic_rank = c("Family", "Genus"),
combine = FALSE
)
plots[["Family"]]
# Multiple ranks as a list
plots <- simple_venn_pq(
data_fungi_mini, "Height",
taxonomic_rank = c("Family", "Genus"),
combine = FALSE
)
plots[["Family"]]
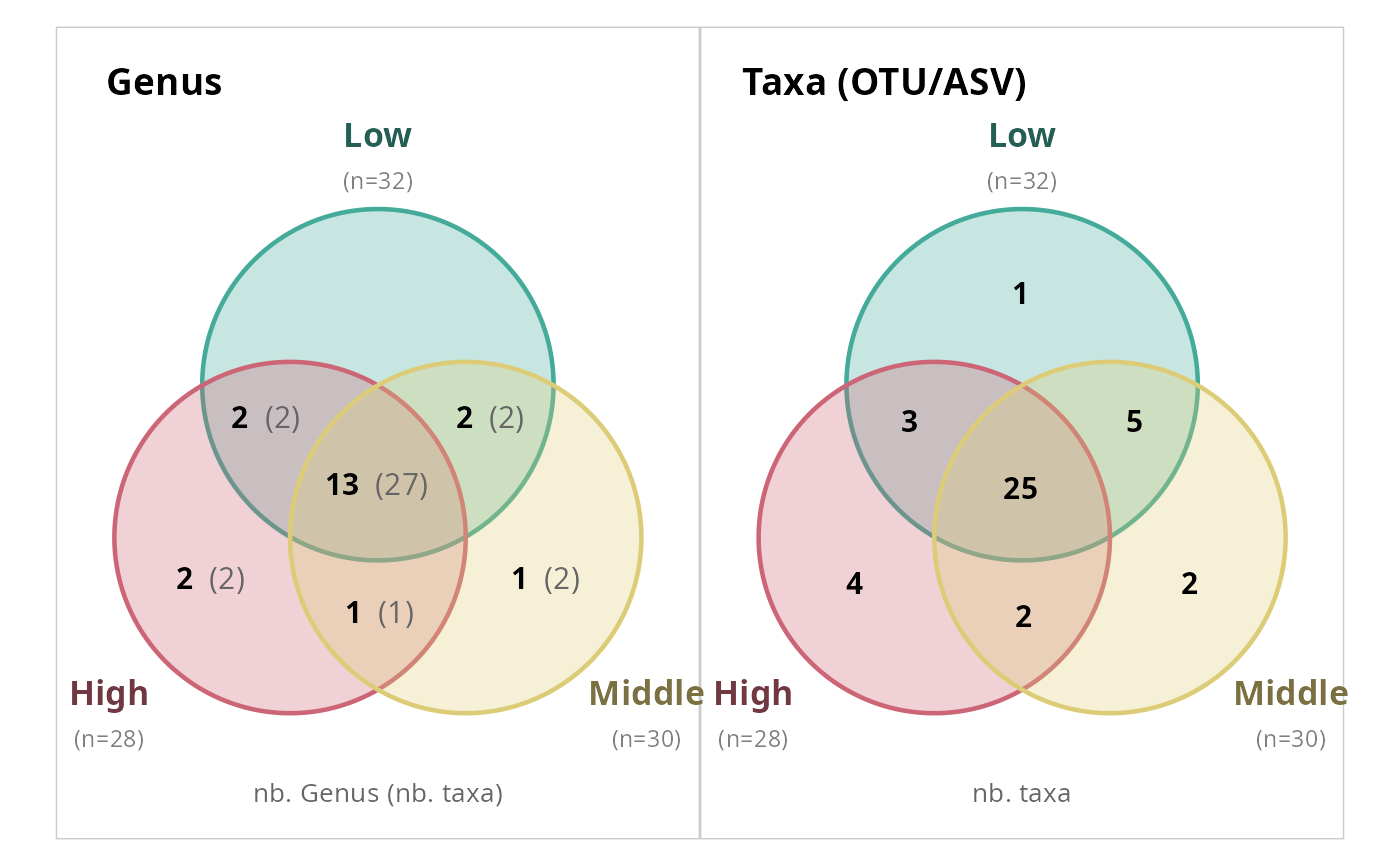
 # Count ASVs instead of rank levels
simple_venn_pq(data_fungi_mini, "Height",
taxonomic_rank = "Genus", count_type = "taxa"
)
# Count ASVs instead of rank levels
simple_venn_pq(data_fungi_mini, "Height",
taxonomic_rank = "Genus", count_type = "taxa"
)
 # Scale text by count value
simple_venn_pq(data_fungi_mini, "Height",
taxonomic_rank = "Genus", scale_text = TRUE
)
# Scale text by count value
simple_venn_pq(data_fungi_mini, "Height",
taxonomic_rank = "Genus", scale_text = TRUE
)
 # From a list_phyloseq object
# Subset to the 80 most abundant taxa to keep the example fast
# (the full data_fungi has 1420 taxa).
data_fungi_small <- prune_taxa(
names(sort(taxa_sums(data_fungi), decreasing = TRUE))[1:80],
data_fungi
)
data_fungi_small <- clean_pq(prune_samples(
sample_sums(data_fungi_small) >= 500, data_fungi_small
))
lpq <- list_phyloseq(list(
fungi = data_fungi_mini,
fungi2 = data_fungi_small
))
#> ℹ Building summary table for 2 phyloseq objects...
#> ℹ Computing comparison characteristics...
#> ℹ Checking sample and taxa overlap...
#> ℹ Detected comparison type: EXPLORATION
#> ℹ 118 common samples, 43 common taxa
#> ✔ list_phyloseq created (EXPLORATION)
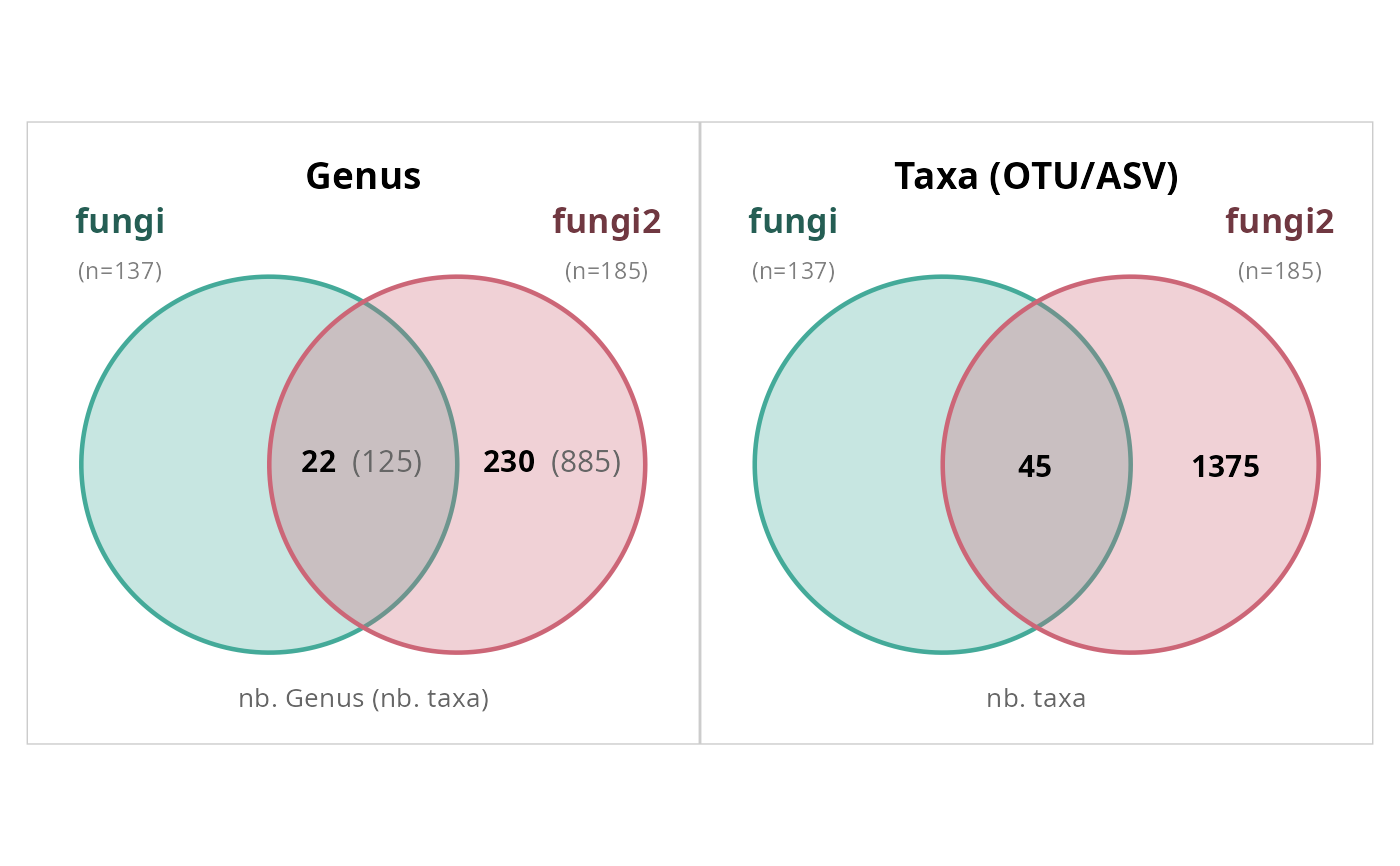
simple_venn_pq(lpq, taxonomic_rank = "Genus")
#> Merging 2 phyloseq objects by refseq: 45 + 80 taxa -> 82 unique sequences.
# From a list_phyloseq object
# Subset to the 80 most abundant taxa to keep the example fast
# (the full data_fungi has 1420 taxa).
data_fungi_small <- prune_taxa(
names(sort(taxa_sums(data_fungi), decreasing = TRUE))[1:80],
data_fungi
)
data_fungi_small <- clean_pq(prune_samples(
sample_sums(data_fungi_small) >= 500, data_fungi_small
))
lpq <- list_phyloseq(list(
fungi = data_fungi_mini,
fungi2 = data_fungi_small
))
#> ℹ Building summary table for 2 phyloseq objects...
#> ℹ Computing comparison characteristics...
#> ℹ Checking sample and taxa overlap...
#> ℹ Detected comparison type: EXPLORATION
#> ℹ 118 common samples, 43 common taxa
#> ✔ list_phyloseq created (EXPLORATION)
simple_venn_pq(lpq, taxonomic_rank = "Genus")
#> Merging 2 phyloseq objects by refseq: 45 + 80 taxa -> 82 unique sequences.
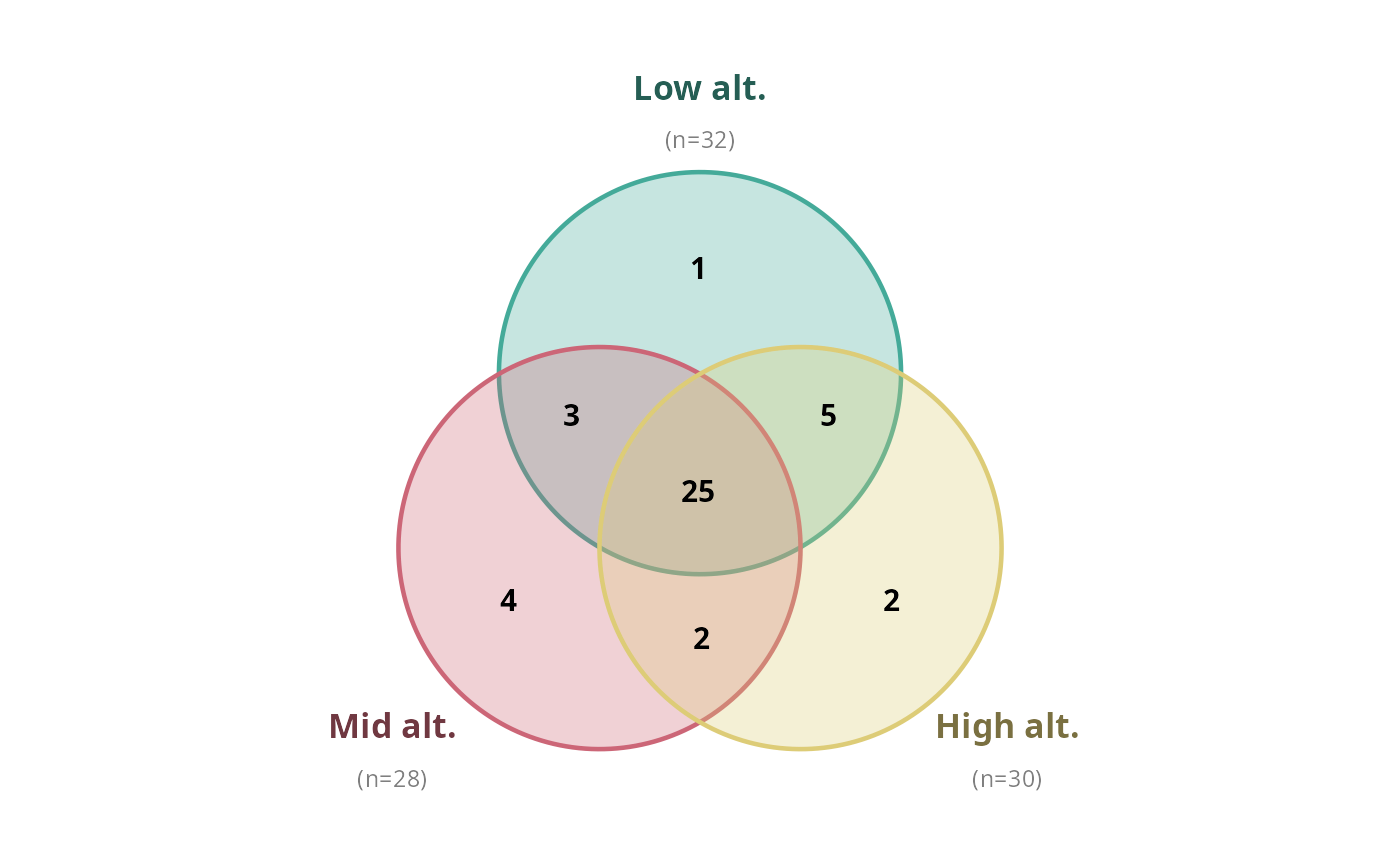
 simple_venn_pq(data_fungi_mini, "Height",
taxonomic_rank = NULL,
labels = c("Low alt.", "Mid alt.", "High alt.")
)
simple_venn_pq(data_fungi_mini, "Height",
taxonomic_rank = NULL,
labels = c("Low alt.", "Mid alt.", "High alt.")
)