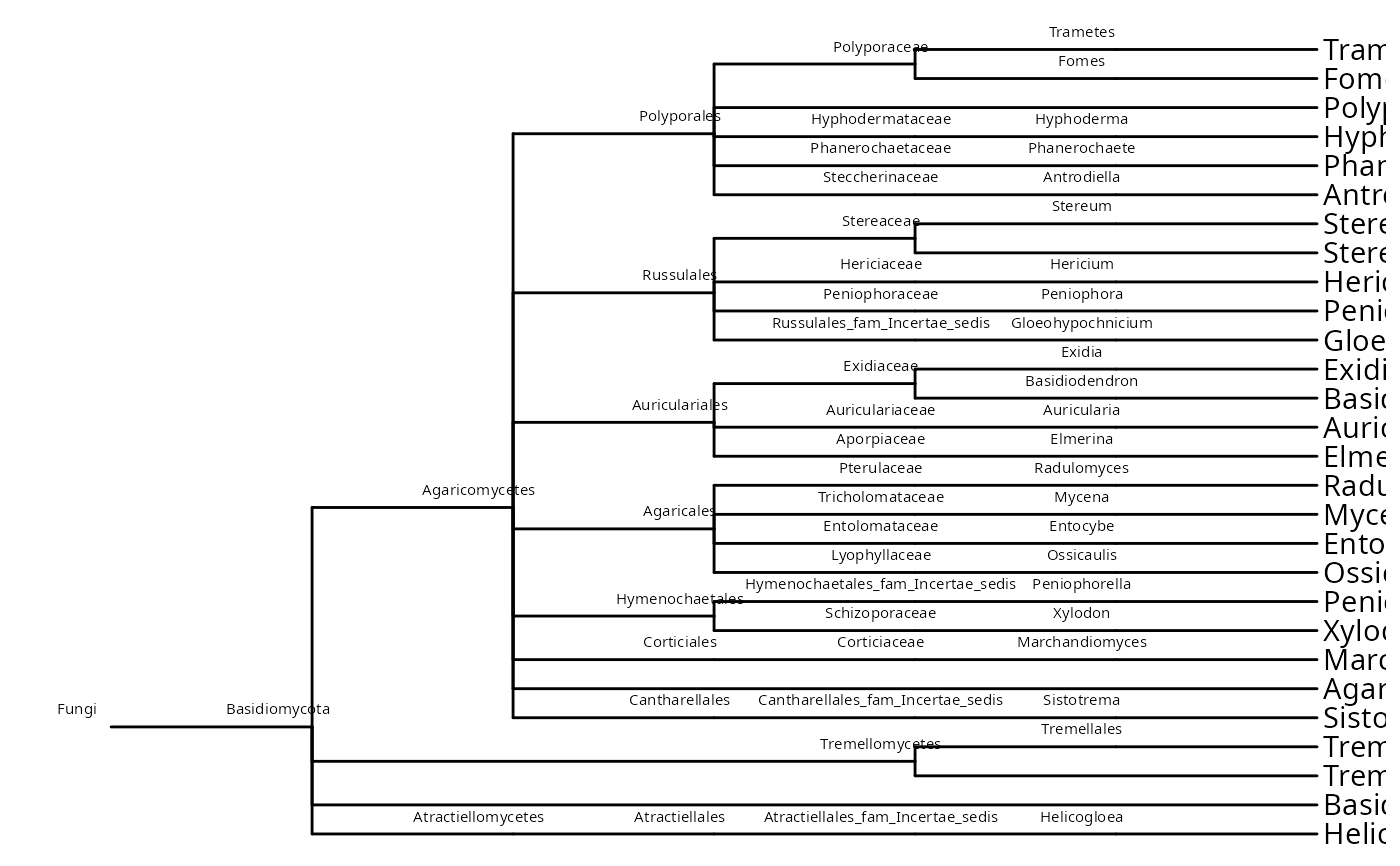
Creates a phylo object from a taxonomy table with hierarchical taxonomic ranks as columns.
Usage
taxo2tree(
physeq,
ranks = c("Domain", "Phylum", "Class", "Order", "Family", "Genus", "Species"),
internal_node_singletons = TRUE,
use_taxa_names = TRUE
)Arguments
- physeq
(required) A
phyloseq-classobject- ranks
Character vector specifying the column names to use as taxonomic ranks, ordered from highest to lowest. By default: c("Domain", "Phylum", "Class", "Order", "Family", "Genus", "Species").
- internal_node_singletons
Logical, if TRUE, create internal nodes for singleton. If FALSE, internal nodes with only one descendant are discarded.
- use_taxa_names
Logical, if TRUE (default), use the taxa names (rownames, e.g., ASV_1, ASV_2) as terminal leaves. If FALSE, collapse identical taxonomy paths and use the lowest rank value as tip labels. This is useful for creating cleaner trees that show only taxonomic structure without individual ASV/OTU names.
Examples
data_fungi_mini@phy_tree <-
phyloseq::phy_tree(taxo2tree(data_fungi_mini,
ranks = c(
"Domain", "Phylum", "Class", "Order",
"Family", "Genus", "Genus_species"
)
))
library(ggtree)
#> ggtree v4.2.0 Learn more at https://yulab-smu.top/contribution-tree-data/
#>
#> Please cite:
#>
#> Shuangbin Xu, Lin Li, Xiao Luo, Meijun Chen, Wenli Tang, Li Zhan, Zehan
#> Dai, Tommy T. Lam, Yi Guan, Guangchuang Yu. Ggtree: A serialized data
#> object for visualization of a phylogenetic tree and annotation data.
#> iMeta 2022, 1(4):e56. doi:10.1002/imt2.56
#>
#> Attaching package: ‘ggtree’
#> The following object is masked from ‘package:MiscMetabar’:
#>
#> multiplot
ggtree(data_fungi_mini@phy_tree) +
geom_nodelab(size = 2, nudge_x = -0.2, nudge_y = 0.6) +
geom_tiplab()
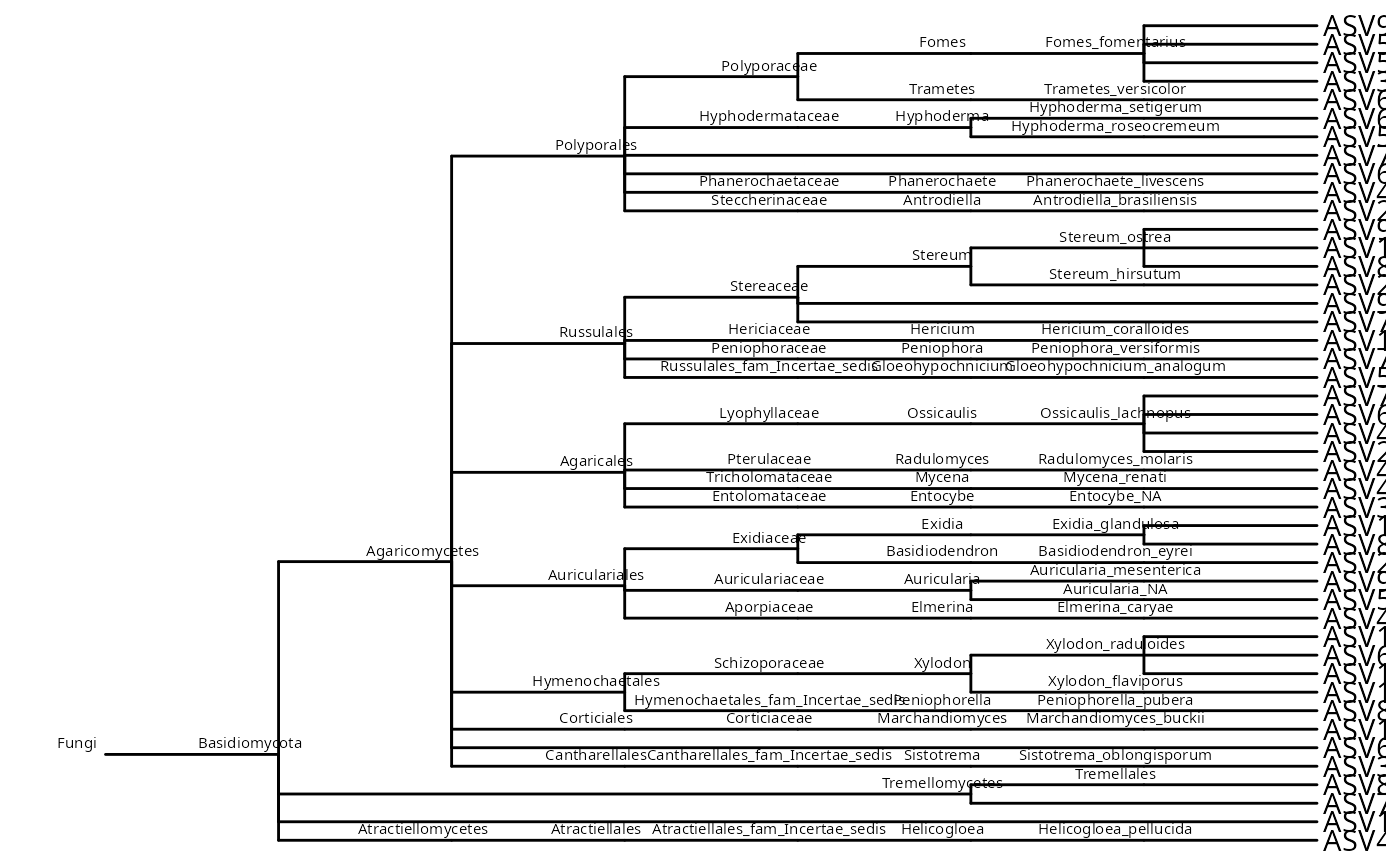 psm <- psmelt(data_fungi_mini) |>
group_by(OTU) |>
summarize(
Mol_Abund = sum(Abundance),
Frequency = sum(Abundance > 0),
Class = tidyr::replace_na(unique(Class), "Unknown")
)
ggtree(data_fungi_mini@phy_tree) %<+% psm +
geom_tiplab(offset = .6, hjust = .5) +
geom_tippoint(aes(
shape = Class,
color = log10(1 + Mol_Abund),
size = Frequency
)) +
geom_nodelab(nudge_x = -0.2, nudge_y = 0.6) +
scale_color_viridis_c()
psm <- psmelt(data_fungi_mini) |>
group_by(OTU) |>
summarize(
Mol_Abund = sum(Abundance),
Frequency = sum(Abundance > 0),
Class = tidyr::replace_na(unique(Class), "Unknown")
)
ggtree(data_fungi_mini@phy_tree) %<+% psm +
geom_tiplab(offset = .6, hjust = .5) +
geom_tippoint(aes(
shape = Class,
color = log10(1 + Mol_Abund),
size = Frequency
)) +
geom_nodelab(nudge_x = -0.2, nudge_y = 0.6) +
scale_color_viridis_c()
 # Without internal node singletons and only until the Genus level
tree_wo_singletons <-
phyloseq::phy_tree(taxo2tree(data_fungi_mini,
ranks = c(
"Domain", "Phylum", "Class", "Order",
"Family", "Genus"
),
internal_node_singletons = FALSE
))
ggtree::ggtree(tree_wo_singletons) +
ggtree::geom_nodelab(size = 2, nudge_x = -0.2, nudge_y = 0.6) +
ggtree::geom_tiplab()
# Without internal node singletons and only until the Genus level
tree_wo_singletons <-
phyloseq::phy_tree(taxo2tree(data_fungi_mini,
ranks = c(
"Domain", "Phylum", "Class", "Order",
"Family", "Genus"
),
internal_node_singletons = FALSE
))
ggtree::ggtree(tree_wo_singletons) +
ggtree::geom_nodelab(size = 2, nudge_x = -0.2, nudge_y = 0.6) +
ggtree::geom_tiplab()
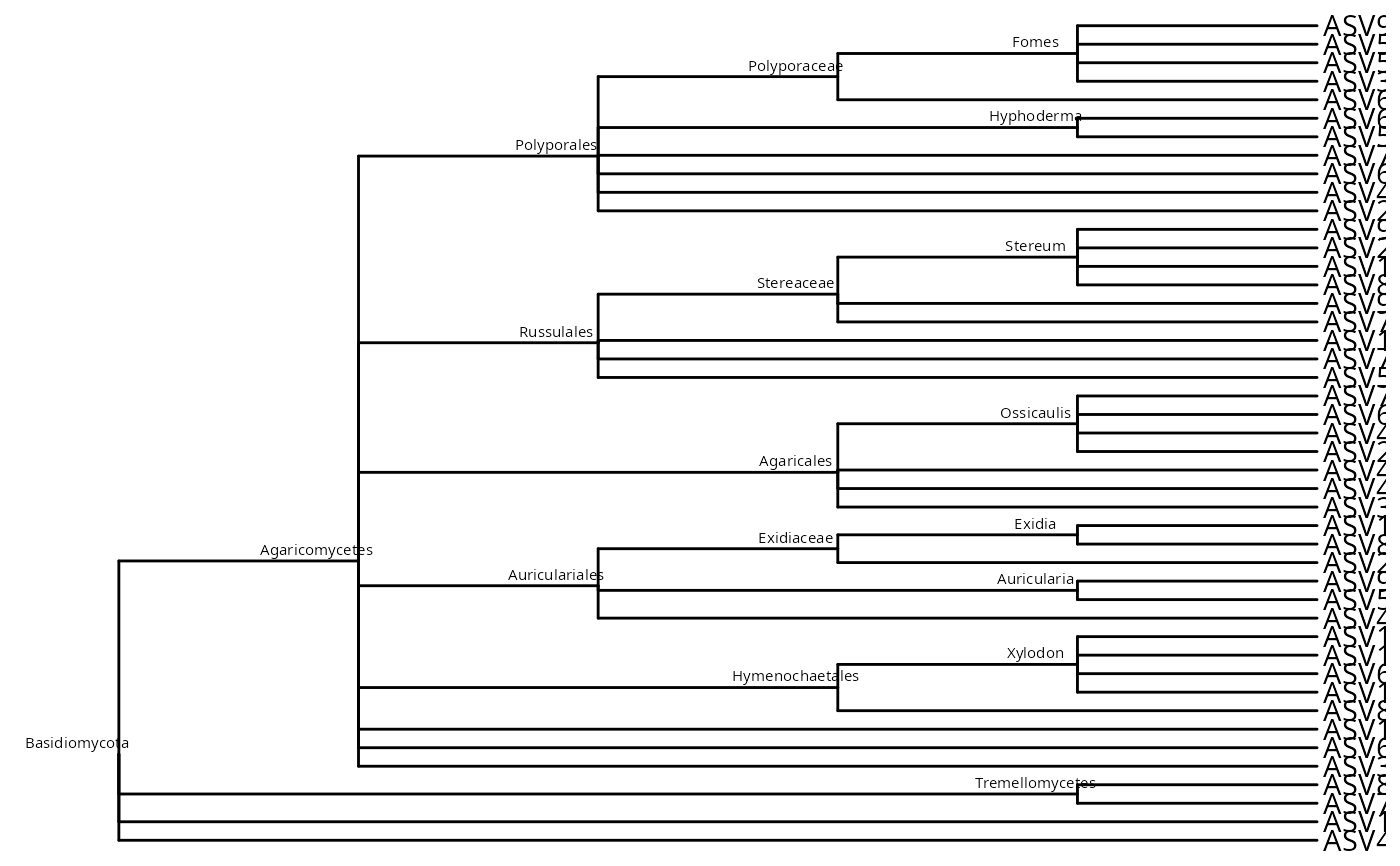 # Without taxa names (collapse identical paths)
tree_no_taxa <- taxo2tree(data_fungi_mini,
ranks = c("Domain", "Phylum", "Class", "Order", "Family", "Genus"),
use_taxa_names = FALSE
)
ggtree::ggtree(tree_no_taxa) +
ggtree::geom_nodelab(size = 2, nudge_x = -0.2, nudge_y = 0.6) +
ggtree::geom_tiplab()
# Without taxa names (collapse identical paths)
tree_no_taxa <- taxo2tree(data_fungi_mini,
ranks = c("Domain", "Phylum", "Class", "Order", "Family", "Genus"),
use_taxa_names = FALSE
)
ggtree::ggtree(tree_no_taxa) +
ggtree::geom_nodelab(size = 2, nudge_x = -0.2, nudge_y = 0.6) +
ggtree::geom_tiplab()