Useful to compare taxonomy from two different source/db/algo side-by-side
Usage
tc_bar(
physeq,
rank_1,
rank_2,
color_rank,
point_size = 0.3,
point_alpha = 0.3,
merge_sample_by = NULL,
log10trans = TRUE
)Arguments
- physeq
(required) A
phyloseq-classobject obtained using thephyloseqpackage.- rank_1
(character or integer) Define the first taxonomic rank as the number or the name of the column in tax_table slot.
- rank_2
(character or integer) Define the second taxonomic rank as the number or the name of the column in tax_table slot.
- color_rank
(character or integer) Define the taxonomic rank for color as the number or the name of the column in tax_table slot.
- point_size
(numeric, default 0.3) Size of points.
- point_alpha
(numeric, default 0.3) Transparency of points.
- merge_sample_by
(character, default NULL) A vector to determine which samples to merge using
merge_samples2()function. Need to be inphyseq@sam_data.- log10trans
(logical, default TRUE) If TRUE, the abundance is log10 transformed.
Examples
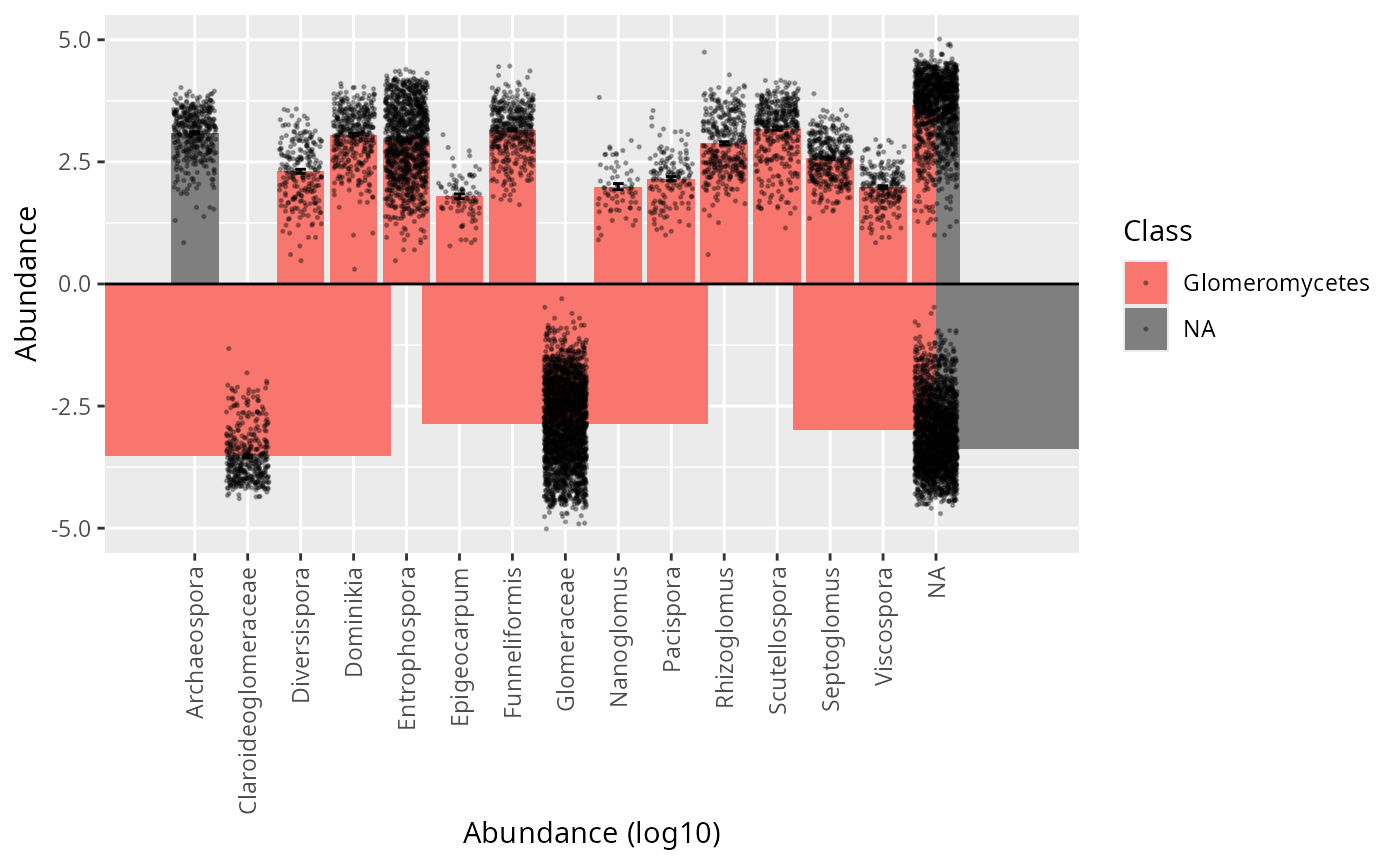
tc_bar(subset_taxa_pq(Glom_otu, taxa_sums(Glom_otu) > 5000), rank_1 = 5, rank_2 = 13, color_rank = 3)
#> Cleaning suppress 0 taxa ( ) and 1 sample(s) ( samp_Blanc-PCR-racines ).
#> Number of non-matching ASV 0
#> Number of matching ASV 1147
#> Number of filtered-out ASV 955
#> Number of kept ASV 192
#> Number of kept samples 443
#> Warning: The `fun.y` argument of `stat_summary()` is deprecated as of ggplot2 3.3.0.
#> ℹ Please use the `fun` argument instead.
#> ℹ The deprecated feature was likely used in the comparpq package.
#> Please report the issue at
#> <https://github.com/adrientaudiere/comparpq/issues>.
 if (FALSE) { # \dontrun{
tc_bar(Glom_otu, rank_1 = 5, rank_2 = 13, color_rank = 3)
tc_bar(as_binary_otu_table(Glom_otu), rank_1 = 5, rank_2 = 13, color_rank = 3, log10trans = FALSE)
tc_bar(Glom_otu,
rank_1 = "Genus",
rank_2 = "Genus__eukaryome_Glomero",
color_rank = "Family"
)
} # }
if (FALSE) { # \dontrun{
tc_bar(Glom_otu, rank_1 = 5, rank_2 = 13, color_rank = 3)
tc_bar(as_binary_otu_table(Glom_otu), rank_1 = 5, rank_2 = 13, color_rank = 3, log10trans = FALSE)
tc_bar(Glom_otu,
rank_1 = "Genus",
rank_2 = "Genus__eukaryome_Glomero",
color_rank = "Family"
)
} # }