dbpq manages FASTA reference databases used for taxonomic assignment in metabarcoding pipelines. It provides tools to download, format, summarize, and modify databases. Part of the pqverse ecosystem.
Anatomy of a reference database
A reference database for metabarcoding taxonomic assignment is characterized by seven key properties:
Name & source — The database name, version, and official website (e.g., SILVA, UNITE). Tracking the exact version used is essential for reproducibility.
Marker — The targeted genetic marker or region (e.g., SSU/18S, LSU/28S, ITS1/ITS2, 16S, COI). A database is generally specific to one or a few markers and primer pairs.
Taxonomic focus — The clade(s) for which the database provides sufficient coverage to enable reliable taxonomic assignment (e.g., Fungi for UNITE, protists for PR2, arbuscular mycorrhizal fungi for MaarjAM). This determines which groups can be assigned with confidence.
Secondary coverage — Sequences from clades outside the primary focus, present in low numbers. These allow reads from non-target organisms to be classified and excluded rather than misassigned (e.g., plant or animal ITS sequences in UNITE). Only a few representative sequences per secondary group are needed; secondary coverage does not enable fine-grained assignment within those groups.
-
Taxonomic format — How taxonomy is encoded in the database:
- Number of files (single annotated FASTA vs. separate sequence + taxonomy files)
- Use of a guide tree for phylogeny-aware classifiers
- Handling of uninformative ranks (e.g., whether
unknown_familyorunclassified_orderare allowed in headers) - Header syntax (SINTAX:
tax=k:Fungi,p:Ascomycota; dada2:k__Fungi;p__Ascomycota; or custom formats)
-
Clustering — Whether sequences are grouped to reduce redundancy, and how:
- Identity-based clustering (e.g., SILVA at 99% sequence identity)
- Biologically meaningful units (e.g., UNITE Species Hypotheses, SH), which collapses intraspecific variation while preserving species-level resolution
-
Filtering — Pre-processing steps applied before or after download:
- Taxonomic scope restriction (e.g., keeping only a target clade to reduce database size)
- Removal of non-informative sequences (e.g., entries assigned only to
unknown_orderthat cannot contribute to assignment) - Sequence trimming or selection (e.g., extracting the ITS region with ITSx, or applying quality scores such as Pintail values in SILVA)
dbpq provides tools to download pre-built databases, convert between header formats, and apply custom filtering steps to address points 5–7.
Installation
# Install from GitHub
# install.packages("pak")
pak::pak("adrientaudiere/dbpq")Quick start
library(dbpq)
# Download UNITE v10.0 for fungi (default: k__/p__ archive)
download_unite_db(dest_dir = "databases")
# Download SILVA SSU dada2-formatted training set
download_silva_db(dest_dir = "databases", format = "dada2")
# Download latest PR2 in dada2 format
download_pr2_db(dest_dir = "databases")
# Download Greengenes2 16S for Bacteria/Archaea
download_greengenes2_db(dest_dir = "databases")
# Download the LTPlus 16S FASTA for Bacteria/Archaea
download_ltplus_db(dest_dir = "databases")Supported databases
Taxonomic coverage overview
Tree of Life
├── Bacteria ──────────────── SILVA (SSU 16S), Greengenes2 (SSU 16S), RDP (SSU 16S), KSGP (SSU 16S), LTPlus (SSU 16S)
├── Archaea ───────────────── SILVA (SSU 16S), Greengenes2 (SSU 16S), KSGP⁴ (SSU 16S), LTPlus (SSU 16S)
└── Eukaryota
├── "Protists"¹ ───────── PR2 (SSU 18S), SILVA (SSU/LSU), Eukaryome (SSU/ITS/LSU)
│ └── incl. plastids ── PR2 (plastid 16S)
├── Fungi ─────────────── UNITE³ (ITS), SILVA (SSU/LSU), Eukaryome (SSU/ITS/LSU), BOLD²
│ └── Glomeromycota ─── MaarjAM (SSU 18S) ← AMF only, Eukaryome (SSU)
├── Viridiplantae ─────── SILVA (SSU/LSU), Eukaryome (SSU/ITS/LSU), BOLD² (matK, rbcL)
├── Metazoa ───────────── SILVA (SSU/LSU), Eukaryome (SSU/LSU), BOLD² (COI), MIDORI2 (COI/12S/16S/Cytb)
└── Organelles ────────── MIDORI2 (mitochondrial 12S/16S/COI/Cytb), Diat.barcode (plastid rbcL)¹ “Protists” is paraphyletic; PR2 covers SAR, Excavata, Amoebozoa, and related unicellular eukaryotic lineages. ² BOLD is query-based: it covers any taxon but only the marker requested. ³ UNITE (eukaryotes version) adds secondary coverage of Viridiplantae and Metazoa (useful for filtering non-fungal reads). ⁴ KSGP includes eukaryote sequences from PR2 and MIDORI2 for contamination control, not for fine-grained eukaryote assignment.
Database details
One row per marker, since each marker targets a different set of organisms. The “Output format” column shows what the download function produces with default parameters.
| Database | Function | Marker | Taxonomic focus | Secondary coverage | Clustering | Output format |
|---|---|---|---|---|---|---|
| UNITE | download_unite_db() |
ITS | Fungi | No | Species Hypotheses (SH) | UNITE k__/p__ (.tgz archive) or SINTAX (.gz) |
| UNITE | download_unite_db(taxon_group = "eukaryotes") |
ITS | Fungi | Yes — Viridiplantae, Metazoa, other eukaryotes | Species Hypotheses (SH) | UNITE k__/p__ (.tgz archive) or SINTAX (.gz) |
| SILVA | download_silva_db() |
SSU 16S/18S | Bacteria, Archaea, Eukaryotes | — | NR99 (99% identity; other versions available directly from SILVA) | dada2 k__/p__ (.fa.gz) |
| SILVA | download_silva_db(target = "LSU") |
LSU 23S/28S | Bacteria, Archaea, Eukaryotes | — | NR99 (99% identity; other versions available directly from SILVA) | Raw SILVA FASTA (.fasta.gz) |
| PR2 | download_pr2_db() |
SSU 18S | Protists | Yes — Metazoa, Fungi, Viridiplantae, organelles | — | dada2 (.fasta.gz) |
| PR2 | download_pr2_db(marker = "plastid") |
Plastid 16S | Plastid-bearing organisms | — | — | dada2 (.fasta.gz) |
| BOLD | download_bold_db(taxon, marker) |
COI-5P, ITS, matK, rbcL, … | All taxa (defined by taxon query) |
— | — | dada2/sintax FASTA (ranked taxonomy from BOLD combined; tax_format) |
| MaarjAM | download_marjaam_db() |
SSU 18S | AMF (Glomeromycota) | No | Virtual Taxa (VT; phylogeny-based) | dada2/sintax FASTA (QIIME release; tax_format) |
| Eukaryome | download_eukaryome_db(url) |
SSU 18S | Eukaryotes | — | — | As-is from URL (dada2 / SINTAX / mothur / QIIME2) |
| Eukaryome | download_eukaryome_db(url) |
ITS | Eukaryotes | — | — | As-is from URL (dada2 / SINTAX / mothur / QIIME2) |
| Eukaryome | download_eukaryome_db(url) |
LSU 28S | Eukaryotes | — | — | As-is from URL (dada2 / SINTAX / mothur / QIIME2) |
| Greengenes2 | download_greengenes2_db() |
SSU 16S | Bacteria, Archaea | — | Reference phylogenetic tree (DEPP placement, not identity-based) | dada2 (d__ prefixes stripped by default; tax_format) (.fa.gz) |
| RDP | download_rdp_db() |
SSU 16S | Bacteria, Archaea | — | Trainset (manually curated) | dada2 k__/p__ (.fa.gz) |
| MIDORI2 | download_midori2_db(url) |
COI, 12S, 16S, Cytb | Metazoa | — | UNIQ (all haplotypes) or LONGEST (one per species) | dada2 / SINTAX / RDP / BLAST (from URL) |
| Diat.barcode | download_diatbarcode_db(url) |
rbcL | Diatoms (Bacillariophyta) | No | Curated (species-level) | dada2 (.fasta.gz) |
| KSGP | download_ksgp_db() |
SSU 16S/18S | Archaea (optimized), Bacteria | Yes — Eukaryota (PR2 + MIDORI2, for contamination control) | None | dada2/sintax FASTA (.tax merged into headers by default via tax_format; partial coverage) |
| KSGP — GTDB+ | download_ksgp_db(database = "GTDB_plus") |
SSU 16S | Bacteria, Archaea | Yes — Eukaryota (PR2 + MIDORI2) | Cleaned (chimera/domain checks) | Plain FASTA (.fasta) + .tax
|
| KSGP — GTDB_cleaned | download_ksgp_db(database = "GTDB_cleaned") |
SSU 16S | Bacteria, Archaea | No | Cleaned (chimera/domain checks) | Plain FASTA (.fasta) + .tax
|
| LTPlus | download_ltplus_db() |
SSU 16S | Bacteria, Archaea | No | Non-redundant (98.7% identity); type strains + curated non-type | 16S FASTA (.fasta, unaligned; DNA + dada2/sintax taxonomy headers by default) |
Database relationships
Databases relate in two distinct ways: some incorporate sequences from others (derivation / nestedness), while others independently cover the same clades (parallel use). The matrix below reads as “sequences from the column database are incorporated into the row database” — every edge verified against each database’s describing paper. Lighter cells mark annotation-only links (taxonomy transferred without sequences).
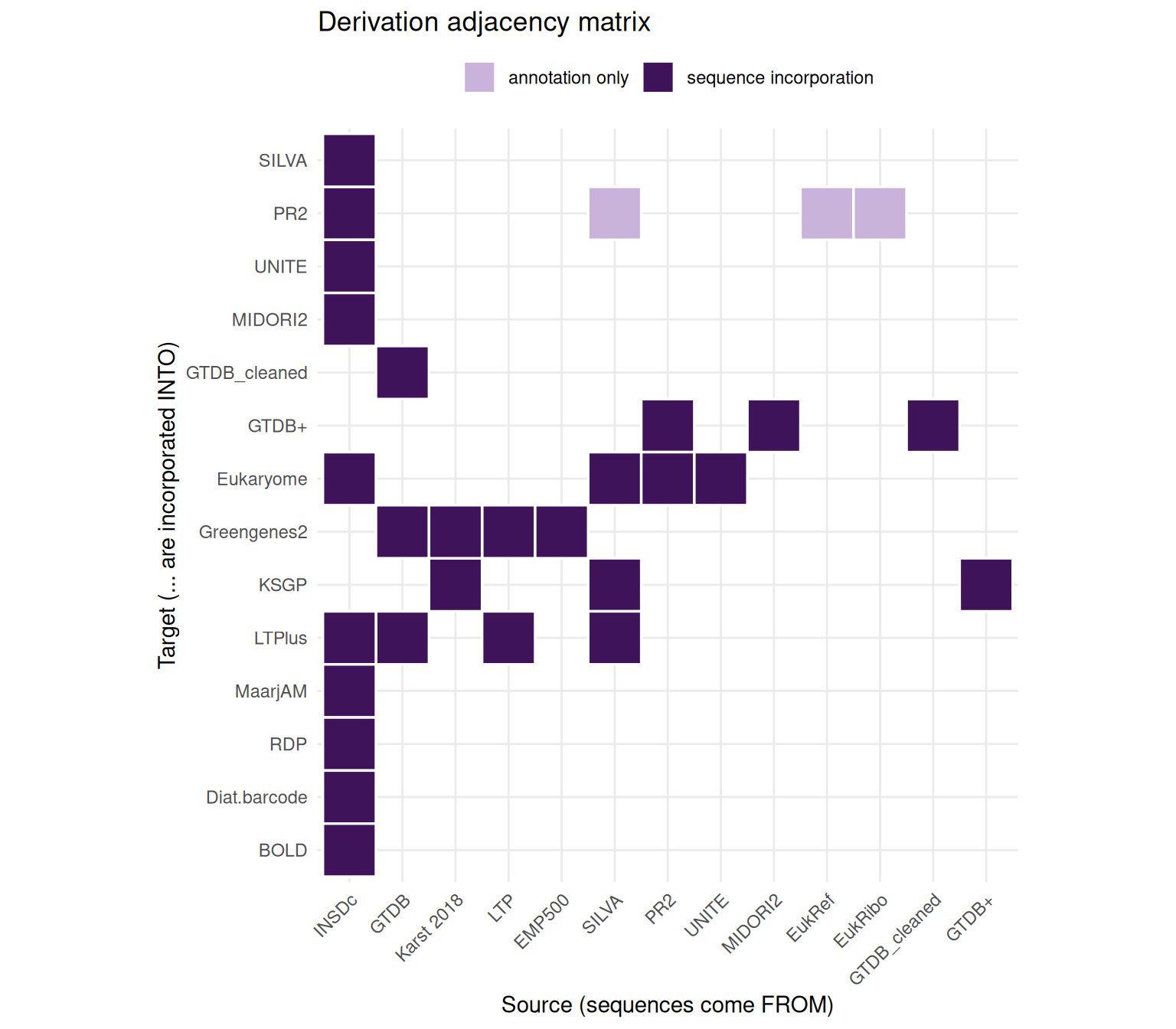
Two relationships are easy to get wrong: Eukaryome is not independent — it compiles sequences from SILVA, PR2 and UNITE — and Greengenes2 derives from GTDB r207 directly (with LTP and Karst 2018), sharing the Karst 2018 source with KSGP.
The full picture — an interactive derivation network, this derivation matrix, and a marker × clade coverage heatmap — is in the article How reference databases relate.
Independent databases — independently curated, with no cross-database sequence incorporation (though all ultimately draw from INSDc/GenBank); use them in parallel based on marker and taxonomic scope:
| Database | Marker | Why independent |
|---|---|---|
| UNITE | ITS | Fungal ITS, distinct marker (but feeds Eukaryome) |
| BOLD | COI, matK, rbcL, … | Barcode markers, independently curated |
| MaarjAM | SSU 18S | AMF-specific, independent curation |
| Diat.barcode | rbcL (plastid) | Diatom-specific, independent curation |
| RDP | SSU 16S | Independent curation, trainset-based |
Navigating the seven properties
The table above documents properties 1–6 per database. The following dbpq functions let you inspect, reformat, recluster, and filter any downloaded database.
Inspect: taxonomic focus & secondary coverage (properties 3–4)
# List all kingdoms or phyla in the database
list_ranks_db("database.fasta", rank_prefix = "k__")
list_ranks_db("database.fasta", rank_prefix = "p__")
# Count sequences from a specific clade (e.g. check secondary coverage)
count_pattern_db("database.fasta", "Viridiplantae")
count_pattern_db("database.fasta", "Metazoa")
# Full overview: sequence count + annotated taxa per rank
summarize_db("database.fasta", tax_format = "auto")Detect & convert format (property 5)
# Auto-detect the taxonomy format of a downloaded file
detect_tax_format("database.fasta")
# Inspect rank prefixes for a known format
tax_prefixes("unite")
tax_prefixes("greengenes2") # uses d__ instead of k__
# Convert to dada2::assignTaxonomy() input
format2dada2(fasta_db = "database.fasta", output_path = "database_dada2.fasta")
# Convert to SINTAX (for VSEARCH --sintax)
format2sintax(fasta_db = "database.fasta", output_path = "database_sintax.fasta")Assess & reduce redundancy (property 6)
# Check database size
count_seq_db("database.fasta")Re-clustering below the downloaded threshold (e.g. from 99% to 97%) requires an external tool:
Filter the database (property 7)
# 7a — Restrict to a taxonomic scope
filter_db("database.fasta", pattern = "Fungi", output = "database_fungi.fasta")
# 7b — Identify non-informative taxa (e.g. "unknown_order", "unclassified")
count_unwanted_tax("database.fasta")
# Keep only sequences with family-level annotation (pattern excludes empty f__ fields)
filter_db("database.fasta", pattern = "f__[A-Za-z]", output = "database_with_family.fasta")
# 7c — Trim primers and select amplicon-length sequences
cutadapt_rm_primers_db(
"database.fasta",
primer_fw = "GCATCGATGAAGAACGCAGC",
primer_rev = "TCCTCCGCTTATTGATATGC",
output = "database_trimmed.fasta"
)Related packages
dbpq is part of the pqverse, a suite of R packages for metabarcoding analysis built around phyloseq objects:
- MiscMetabar — Miscellaneous functions for metabarcoding analyses
- tidypq — Tidyverse-style verbs for phyloseq objects
- comparpq — Compare multiple phyloseq objects
- taxinfo — Taxonomy-based information from GBIF, Wikipedia, GloBI