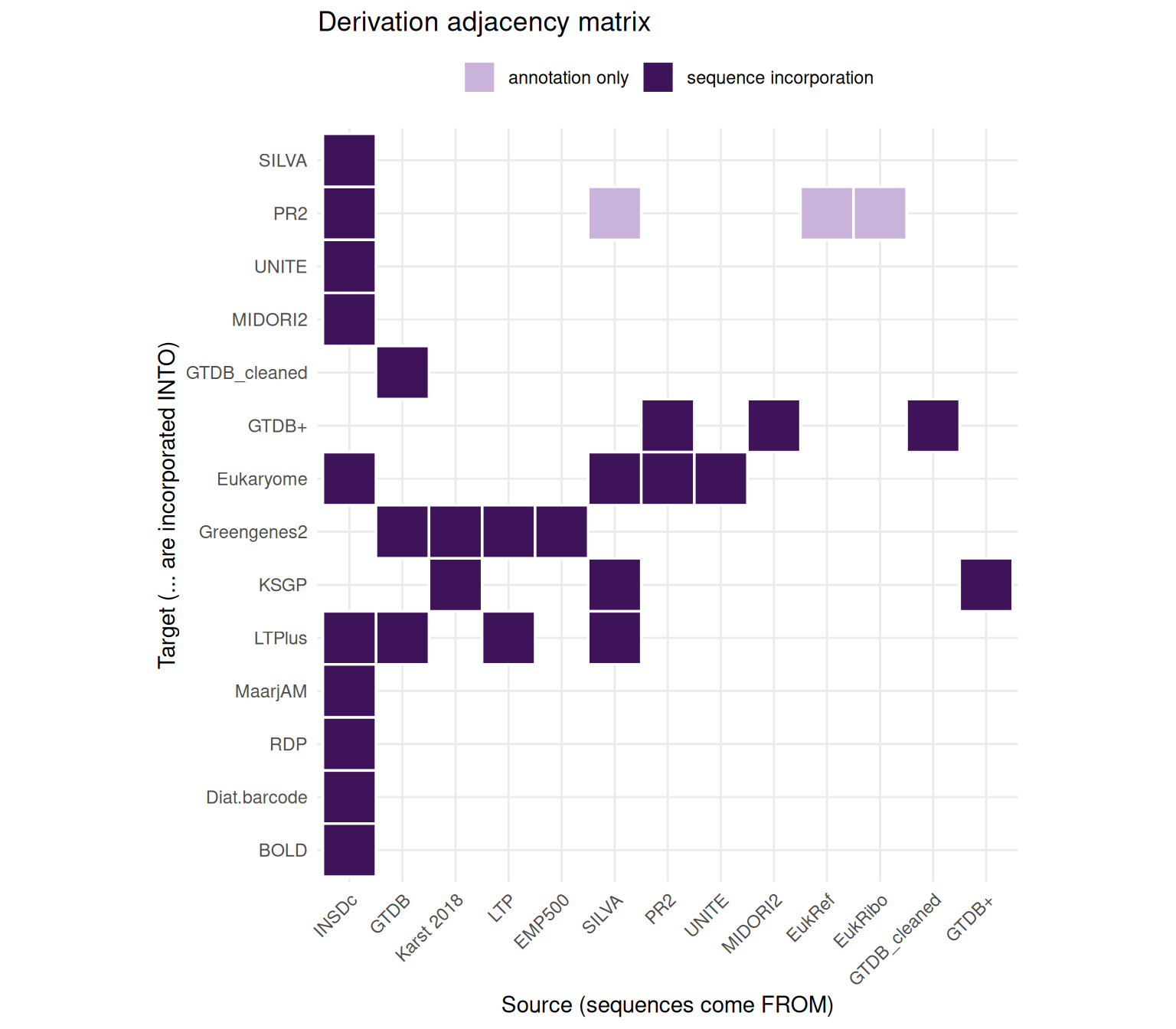
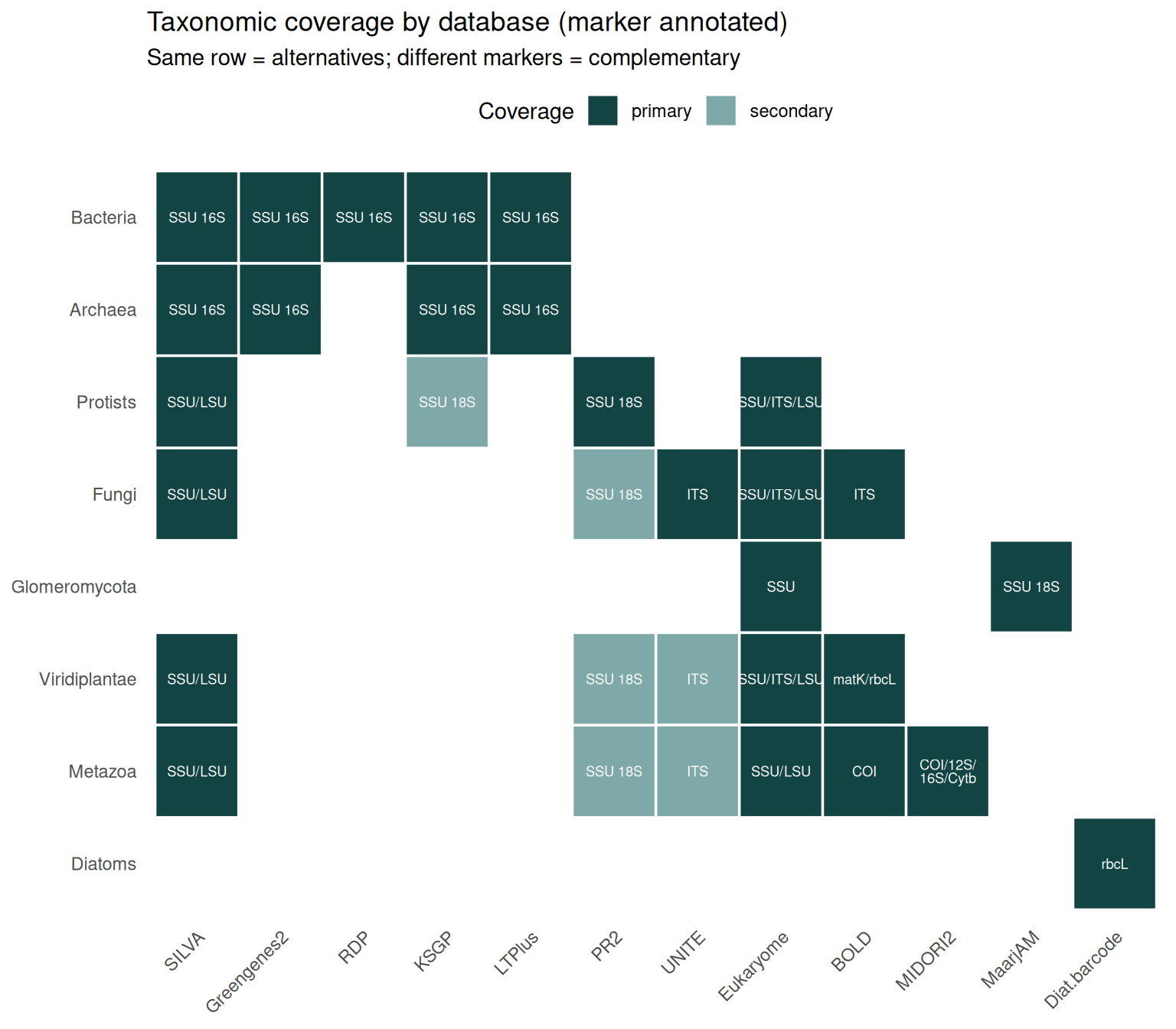
coverage <- tribble(
~database, ~clade, ~marker, ~role,
"SILVA", "Bacteria", "SSU 16S", "primary",
"Greengenes2", "Bacteria", "SSU 16S", "primary",
"RDP", "Bacteria", "SSU 16S", "primary",
"KSGP", "Bacteria", "SSU 16S", "primary",
"LTPlus", "Bacteria", "SSU 16S", "primary",
"SILVA", "Archaea", "SSU 16S", "primary",
"Greengenes2", "Archaea", "SSU 16S", "primary",
"KSGP", "Archaea", "SSU 16S", "primary",
"LTPlus", "Archaea", "SSU 16S", "primary",
"PR2", "Protists", "SSU 18S", "primary",
"SILVA", "Protists", "SSU/LSU", "primary",
"Eukaryome", "Protists", "SSU/ITS/LSU", "primary",
"UNITE", "Fungi", "ITS", "primary",
"SILVA", "Fungi", "SSU/LSU", "primary",
"Eukaryome", "Fungi", "SSU/ITS/LSU", "primary",
"BOLD", "Fungi", "ITS", "primary",
"MaarjAM", "Glomeromycota", "SSU 18S", "primary",
"Eukaryome", "Glomeromycota", "SSU", "primary",
"SILVA", "Viridiplantae", "SSU/LSU", "primary",
"Eukaryome", "Viridiplantae", "SSU/ITS/LSU", "primary",
"BOLD", "Viridiplantae", "matK/rbcL", "primary",
"SILVA", "Metazoa", "SSU/LSU", "primary",
"Eukaryome", "Metazoa", "SSU/LSU", "primary",
"BOLD", "Metazoa", "COI", "primary",
"MIDORI2", "Metazoa", "COI/12S/\n16S/Cytb", "primary",
"Diat.barcode", "Diatoms", "rbcL", "primary",
# secondary (contamination-filtering) coverage
"UNITE", "Viridiplantae", "ITS", "secondary",
"UNITE", "Metazoa", "ITS", "secondary",
"PR2", "Metazoa", "SSU 18S", "secondary",
"PR2", "Fungi", "SSU 18S", "secondary",
"PR2", "Viridiplantae", "SSU 18S", "secondary",
"KSGP", "Protists", "SSU 18S", "secondary"
)
clade_lvls <- c(
"Bacteria", "Archaea", "Protists", "Fungi", "Glomeromycota",
"Viridiplantae", "Metazoa", "Diatoms"
)
db_lvls <- c(
"SILVA", "Greengenes2", "RDP", "KSGP", "LTPlus", "PR2", "UNITE",
"Eukaryome", "BOLD", "MIDORI2", "MaarjAM", "Diat.barcode"
)
coverage <- coverage |>
mutate(
clade = factor(clade, levels = rev(clade_lvls)),
database = factor(database, levels = db_lvls)
)
ggplot(coverage, aes(x = database, y = clade)) +
geom_tile(aes(fill = role), color = "white", linewidth = 0.6) +
geom_text(aes(label = marker), size = 2.4, color = "white", lineheight = 0.8) +
scale_fill_manual(
values = c("primary" = "#124444", "secondary" = "#7fa8a8"),
name = "Coverage"
) +
labs(
x = NULL, y = NULL,
title = "Taxonomic coverage by database (marker annotated)",
subtitle = "Same row = alternatives; different markers = complementary"
) +
theme_minimal(base_size = 11) +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
panel.grid = element_blank(),
legend.position = "top"
)