Creates ggplot2 versions of the four ALDEx2 diagnostic plot types:
MW (Bland-Altman style), MA, volcano, and volcano.var. Supports
faceting by a name column, making it suitable for output from
aldex_lpq().
Arguments
- x
(data.frame or tibble, required) ALDEx2 results, typically from
ALDEx2::aldex()oraldex_lpq(). Must contain columnsdiff.btw,diff.win,rab.all, andeffect. Whentestis"welch", columnwe.eBHis required; when"wilcox", columnwi.eBHis required.- type
(character, default "MW") Plot type. One of
"MW"(dispersion vs difference),"MA"(abundance vs difference),"volcano"(difference vs -log10 q-value), or"volcano.var"(dispersion vs -log10 q-value).- test
(character, default "welch") Statistical test used for significance calling. One of
"welch","wilcox","effect", or"both"(effect + welch).- cutoff.pval
(numeric, default 0.05) q-value threshold for significance.
- cutoff.effect
(numeric, default 1) Effect size threshold (must be >= 0.5 when
testis"effect"or"both").- rare
(numeric, default 0) Abundance threshold below which taxa are marked as rare (only used for MW and MA plots).
- all.col
(character, default "grey30") Color for non-significant points.
- called.col
(character, default "red") Color for significant points.
- rare.col
(character, default "black") Color for rare taxa points.
- point.size
(numeric, default 1.5) Size of points.
- point.alpha
(numeric, default 0.4) Alpha transparency of non-significant points.
Value
A ggplot2 object. If x contains a name column (e.g.,
from aldex_lpq()), the plot is faceted by name.
Details
This function reimplements ALDEx2::aldex.plot() using ggplot2,
providing a more customizable and composable output. The four plot
types correspond to the original ALDEx2 types:
- MW
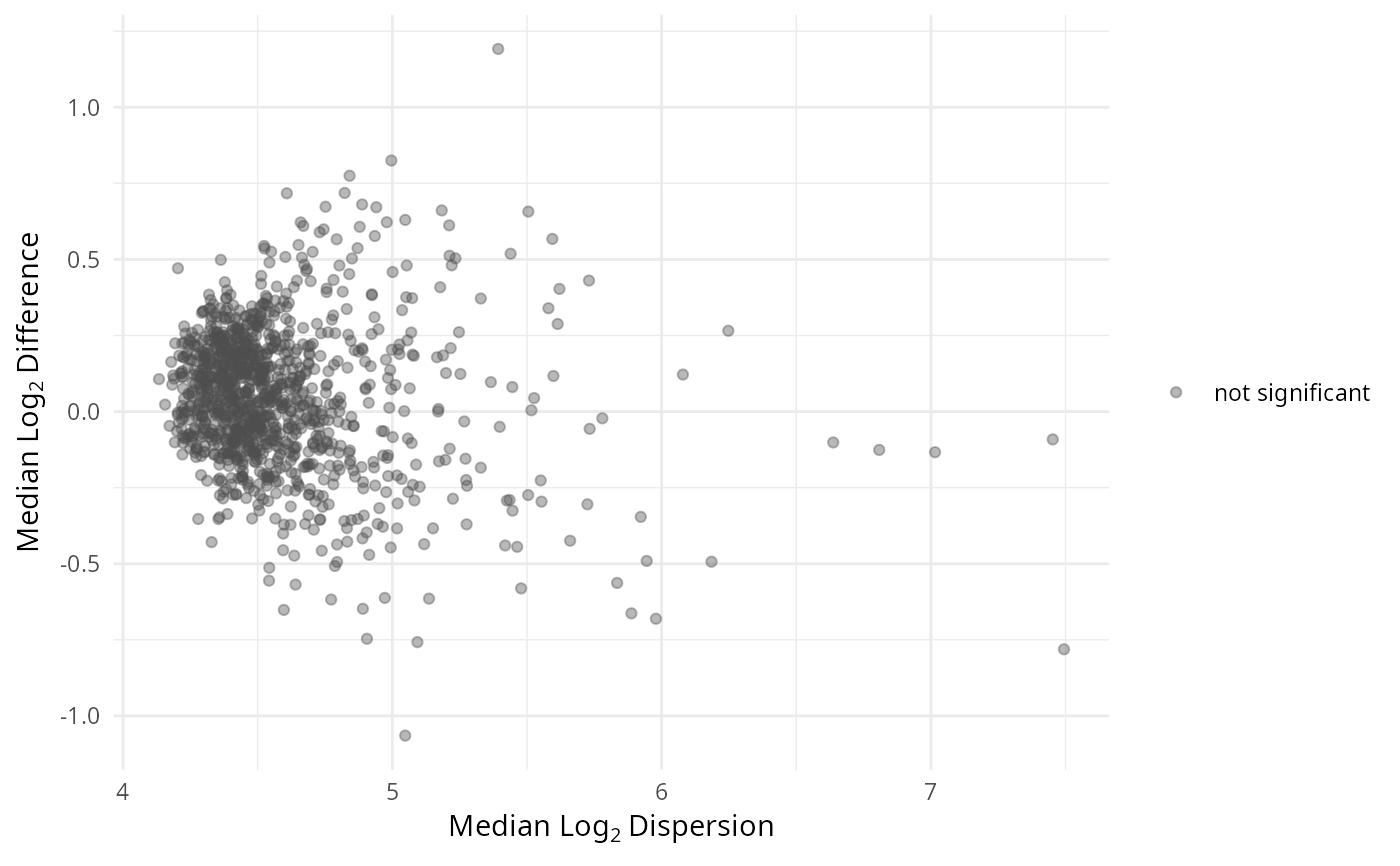
Bland-Altman style: within-condition dispersion (x) vs between-condition difference (y), with +/-1 effect size lines.
- MA
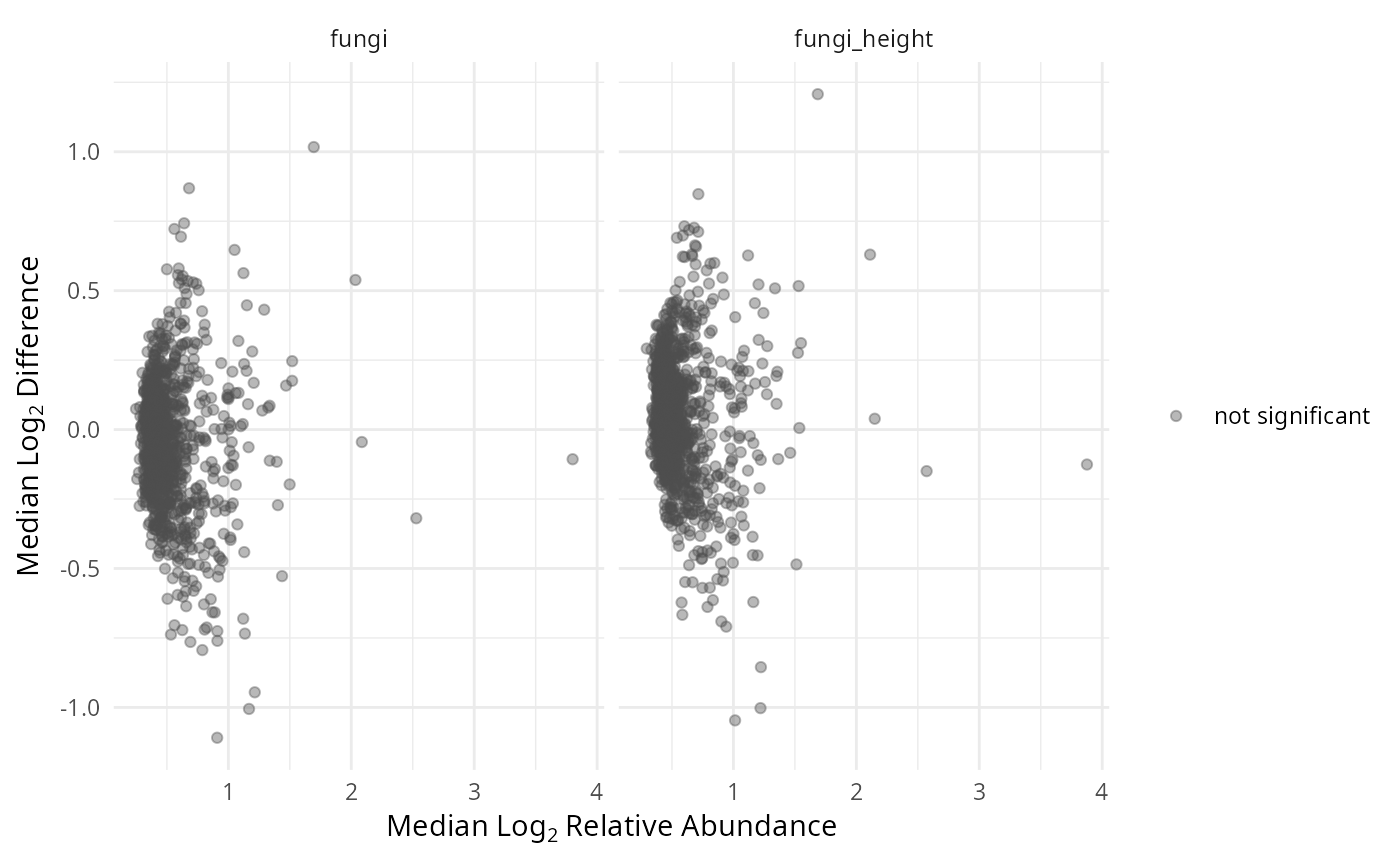
MA plot: median log2 relative abundance (x) vs median log2 difference (y).
- volcano
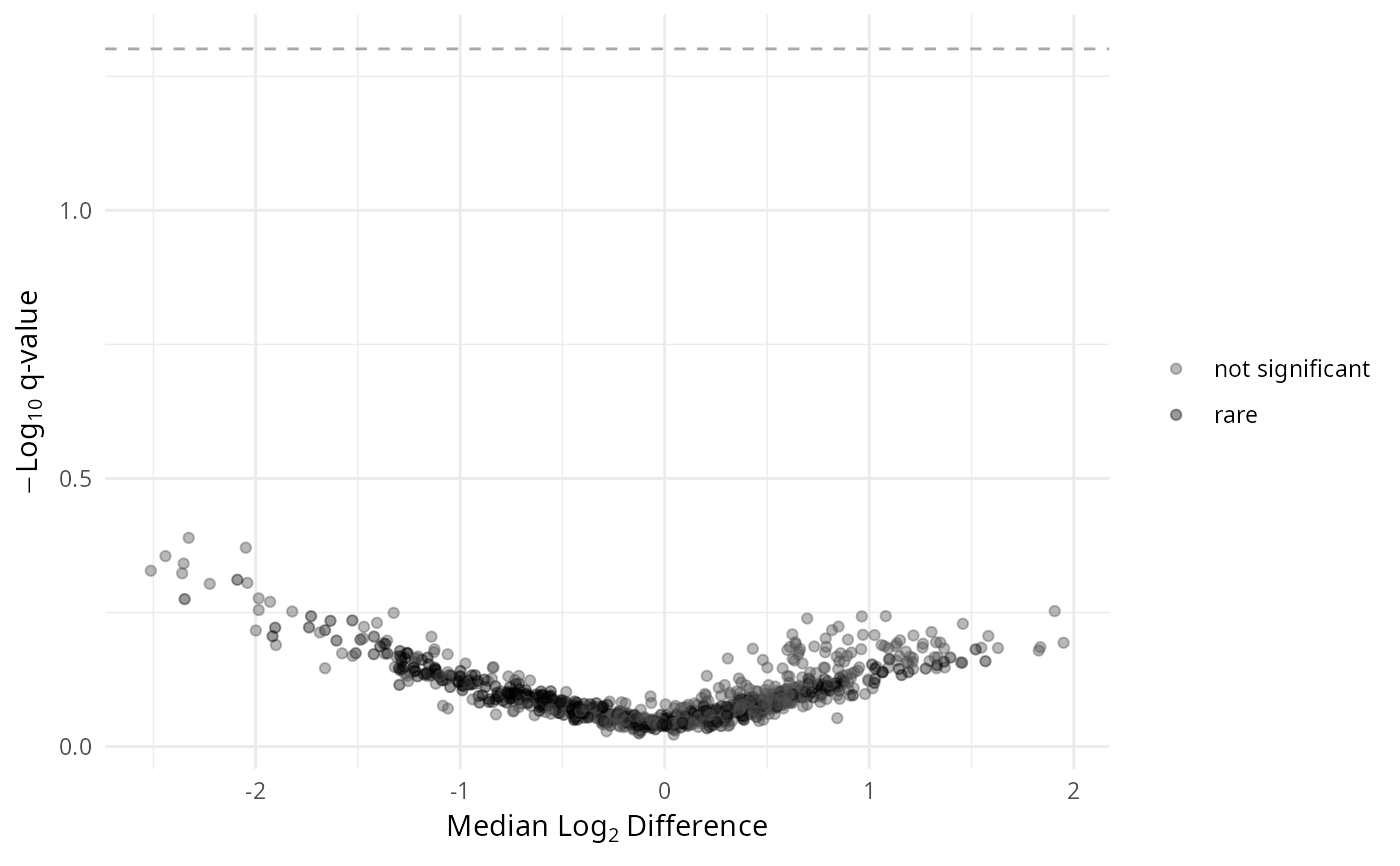
Volcano plot: median log2 difference (x) vs -log10 q-value (y).
- volcano.var
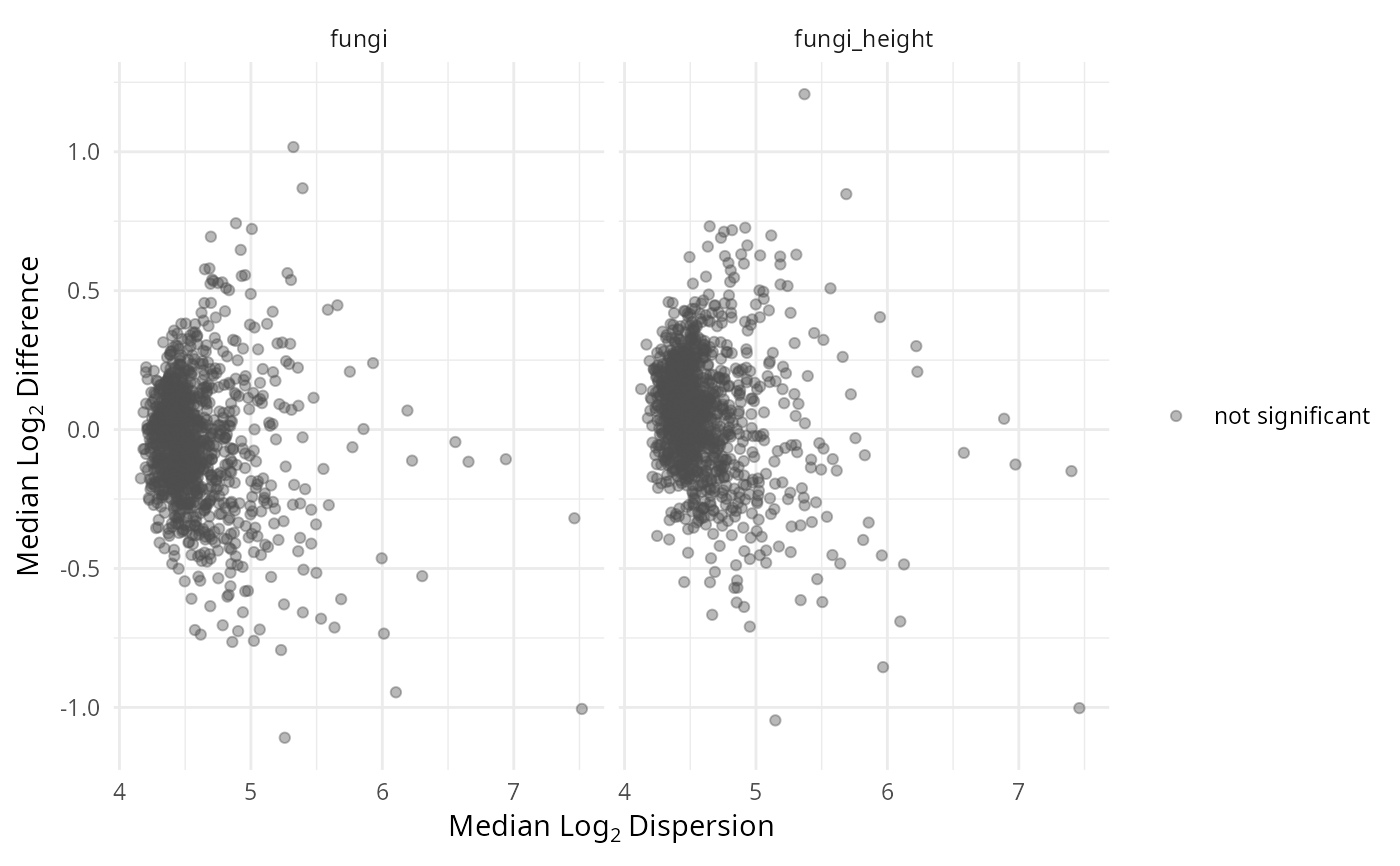
Variance volcano: median log2 dispersion (x) vs -log10 q-value (y).
Examples
# Subset to the 80 most abundant taxa to keep the example fast
# (the full data_fungi has 1420 taxa, which is slow for ALDEx2).
data_fungi_small <- prune_taxa(
names(sort(taxa_sums(data_fungi), decreasing = TRUE))[1:80],
data_fungi
)
data_fungi_small <- clean_pq(prune_samples(
sample_sums(data_fungi_small) >= 500, data_fungi_small
))
data_fungi_high <- multiply_counts_pq(data_fungi_small, "Height", "High",
4,
prop_taxa = 0.1, seed = 42
)
#> Modified 8 taxa in 28 matched samples
aldex_pq(data_fungi_high,
bifactor = "Height",
modalities = c("Low", "High")
) |>
gg_aldex_plot()
#> Taxa are now in rows.
#> aldex.clr: generating Monte-Carlo instances and clr values
#> conditions vector supplied
#> operating in serial mode
#> aldex.scaleSim: adjusting samples to reflect scale uncertainty.
#> aldex.ttest: doing t-test
#> aldex.effect: calculating effect sizes
 lpq <- list_phyloseq(
list(
fungi = data_fungi_small,
fungi_height = data_fungi_high
),
same_bioinfo_pipeline = FALSE
)
#> ℹ Building summary table for 2 phyloseq objects...
#> ℹ Computing comparison characteristics...
#> ℹ Checking sample and taxa overlap...
#> ℹ Detected comparison type: ROBUSTNESS
#> ℹ 127 common samples, 80 common taxa
#> ✔ list_phyloseq created (ROBUSTNESS)
# From aldex_lpq (faceted by name)
lpq_res <- aldex_lpq(lpq,
bifactor = "Height",
modalities = c("Low", "High")
)
#> Running ALDEx2 on 2 phyloseq objects
#> Bifactor: Height
#> → Processing: fungi
#> Taxa are now in rows.
#> aldex.clr: generating Monte-Carlo instances and clr values
#> conditions vector supplied
#> operating in serial mode
#> aldex.scaleSim: adjusting samples to reflect scale uncertainty.
#> aldex.ttest: doing t-test
#> aldex.effect: calculating effect sizes
#> → Processing: fungi_height
#> Taxa are now in rows.
#> aldex.clr: generating Monte-Carlo instances and clr values
#> conditions vector supplied
#> operating in serial mode
#> aldex.scaleSim: adjusting samples to reflect scale uncertainty.
#> aldex.ttest: doing t-test
#> aldex.effect: calculating effect sizes
gg_aldex_plot(lpq_res, type = "MA")
lpq <- list_phyloseq(
list(
fungi = data_fungi_small,
fungi_height = data_fungi_high
),
same_bioinfo_pipeline = FALSE
)
#> ℹ Building summary table for 2 phyloseq objects...
#> ℹ Computing comparison characteristics...
#> ℹ Checking sample and taxa overlap...
#> ℹ Detected comparison type: ROBUSTNESS
#> ℹ 127 common samples, 80 common taxa
#> ✔ list_phyloseq created (ROBUSTNESS)
# From aldex_lpq (faceted by name)
lpq_res <- aldex_lpq(lpq,
bifactor = "Height",
modalities = c("Low", "High")
)
#> Running ALDEx2 on 2 phyloseq objects
#> Bifactor: Height
#> → Processing: fungi
#> Taxa are now in rows.
#> aldex.clr: generating Monte-Carlo instances and clr values
#> conditions vector supplied
#> operating in serial mode
#> aldex.scaleSim: adjusting samples to reflect scale uncertainty.
#> aldex.ttest: doing t-test
#> aldex.effect: calculating effect sizes
#> → Processing: fungi_height
#> Taxa are now in rows.
#> aldex.clr: generating Monte-Carlo instances and clr values
#> conditions vector supplied
#> operating in serial mode
#> aldex.scaleSim: adjusting samples to reflect scale uncertainty.
#> aldex.ttest: doing t-test
#> aldex.effect: calculating effect sizes
gg_aldex_plot(lpq_res, type = "MA")
 gg_aldex_plot(lpq_res, type = "MW", test = "wilcox")
gg_aldex_plot(lpq_res, type = "MW", test = "wilcox")
 gg_aldex_plot(lpq_res, type = "volcano")
gg_aldex_plot(lpq_res, type = "volcano")
 gg_aldex_plot(lpq_res, type = "volcano.var")
gg_aldex_plot(lpq_res, type = "volcano.var")
 # Volcano plot from a single phyloseq object
aldex_res <- aldex_pq(data_fungi_high,
bifactor = "Height",
modalities = c("Low", "High")
)
#> Taxa are now in rows.
#> aldex.clr: generating Monte-Carlo instances and clr values
#> conditions vector supplied
#> operating in serial mode
#> aldex.scaleSim: adjusting samples to reflect scale uncertainty.
#> aldex.ttest: doing t-test
#> aldex.effect: calculating effect sizes
gg_aldex_plot(aldex_res, type = "volcano")
# Volcano plot from a single phyloseq object
aldex_res <- aldex_pq(data_fungi_high,
bifactor = "Height",
modalities = c("Low", "High")
)
#> Taxa are now in rows.
#> aldex.clr: generating Monte-Carlo instances and clr values
#> conditions vector supplied
#> operating in serial mode
#> aldex.scaleSim: adjusting samples to reflect scale uncertainty.
#> aldex.ttest: doing t-test
#> aldex.effect: calculating effect sizes
gg_aldex_plot(aldex_res, type = "volcano")