Run LEfSe on a phyloseq object
Usage
lefser_pq(
physeq,
bifactor = NULL,
modalities = NULL,
compute_relativeAb = TRUE,
by_clade = FALSE,
...
)Arguments
- physeq
(required) a
phyloseq-classobject obtained using thephyloseqpackage.- bifactor
(required) The name of a column present in the
@sam_dataslot of the physeq object. Must be a character vector or a factor.- modalities
(default NULL) A vector of modalities to keep in the analysis. If NULL, all modalities present in bifactor are kept. Note that only two modalities are allowed.
- compute_relativeAb
(logical, default TRUE) Do we compute relative abundance before running LEfSe?
- by_clade
(logical, default FALSE) Do we use the lefserClades function (which test for different depth in the taxonomic classification) or the lefser function (taxa-level)?
- ...
Additional arguments passed on to
lefser::lefser()
Details
It is a wrapper of the lefser::lefser() and lefser::lefserClades() functions.
Examples
# \donttest{
if (requireNamespace("lefser") && requireNamespace("mia")) {
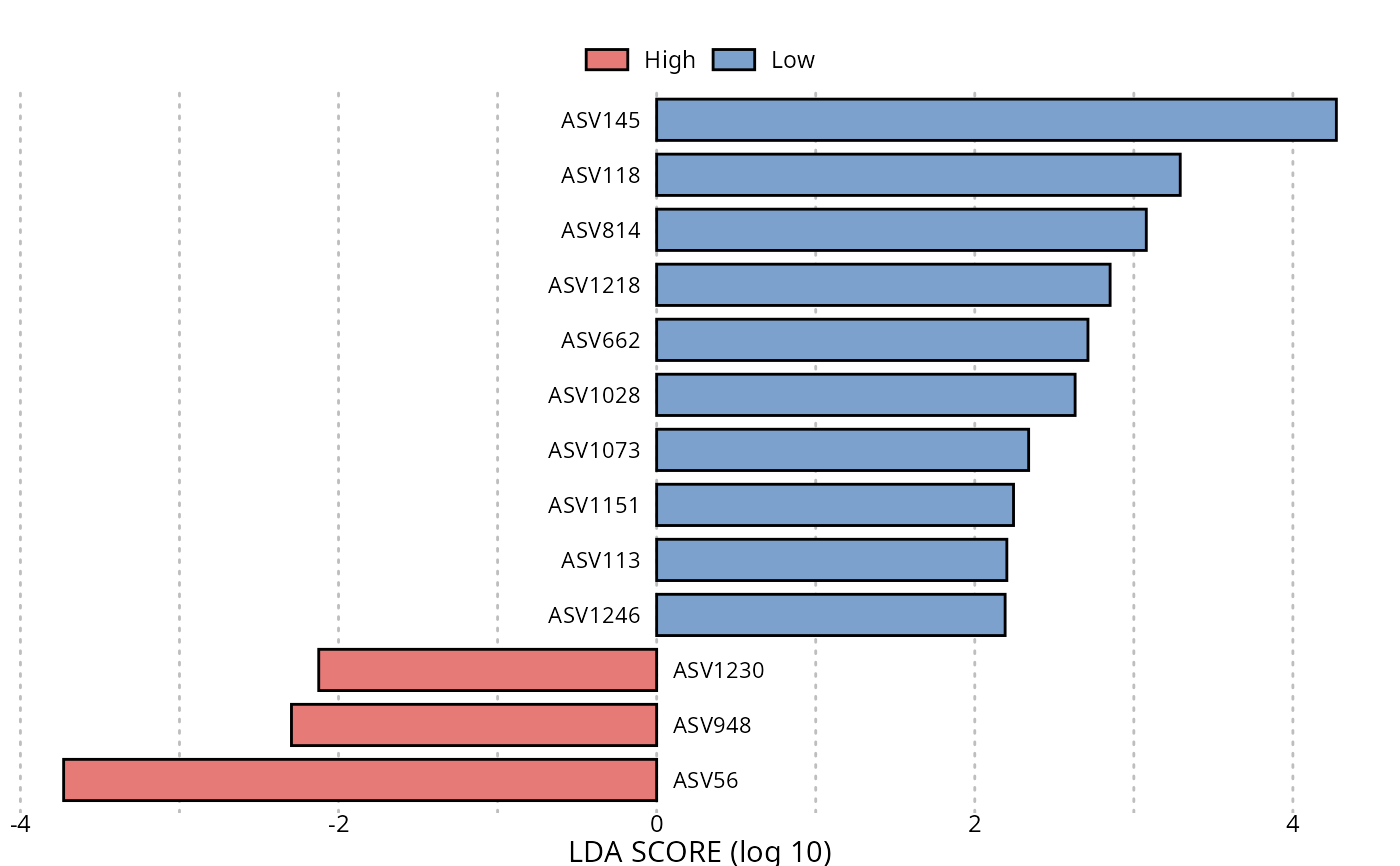
res_lefse <- lefser_pq(data_fungi_mini,
bifactor = "Height",
modalities = c("Low", "High")
)
lefser::lefserPlot(res_lefse)
}
#> Loading required namespace: lefser
#> Registered S3 method overwritten by 'ggtree':
#> method from
#> fortify.igraph ggnetwork
#> Loading required namespace: mia
#> The outcome variable is specified as 'Height' and the reference category is 'High'.
#> See `?factor` or `?relevel` to change the reference category.
#> No significant features found.
 # }
# }