It is a wrapper of the function plot_refseq_extremity_pq(). See
?plot_refseq_extremity_pq for more examples.
If hill_scale is not null, Hill diversity number are used to represent the distribution
of the diversity (equitability) along the sequences.
Usage
plot_refseq_pq(
physeq,
q = NULL,
first_n = min(Biostrings::width(physeq@refseq)),
last_n = NULL,
min_width = first_n
)Arguments
- physeq
(required) a
phyloseq-classobject obtained using thephyloseqpackage.- q
(vector) A vector defining the Hill number wanted. Set to NULL if you don't want to plot Hill diversity metrics. Hill numbers are more appropriate in DNA metabarcoding studies when
q > 0(Alberdi & Gilbert, 2019; Calderón-Sanou et al., 2019).- first_n
(int, default 10) The number of nucleotides to plot the 5' extremity.
- last_n
(int, default 10) The number of nucleotides to plot the 3' extremity.
- min_width
(int, default 0) Select only the sequences from physeq@refseq with using a minimum length threshold. If
first_nis superior to the minimum length of the references sequences, you must use min_width to filter out the narrower sequences
Examples
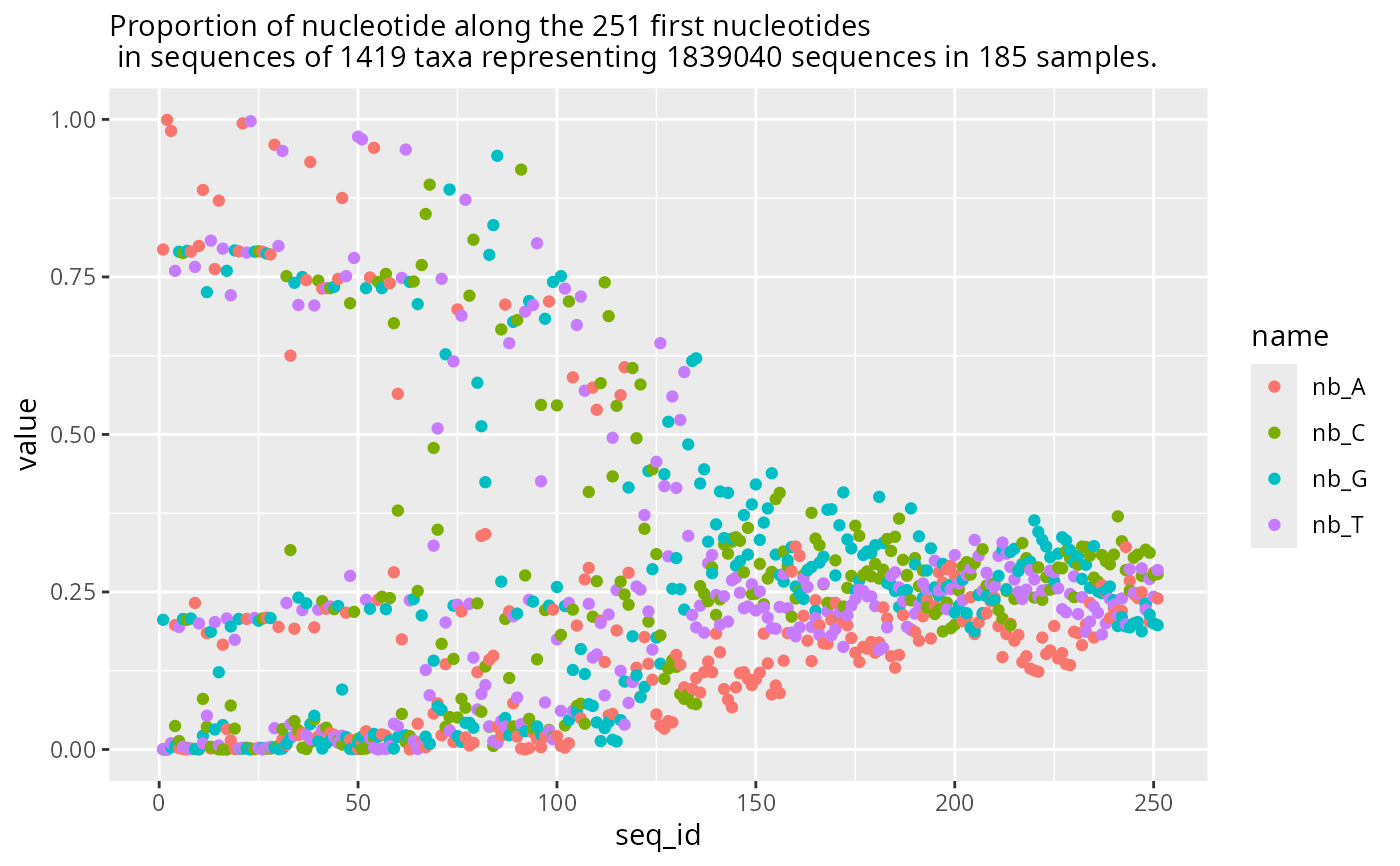
plot_refseq_pq(data_fungi_mini)
#> Cleaning suppress 0 taxa ( ) and 1 sample(s) ( W26-001-B_S165_MERGED.fastq.gz ).
#> Number of non-matching ASV 0
#> Number of matching ASV 45
#> Number of filtered-out ASV 1
#> Number of kept ASV 44
#> Number of kept samples 136
#> Warning: Removed 336 rows containing missing values or values outside the scale range
#> (`geom_point()`).
 if (FALSE) { # \dontrun{
plot_refseq_pq(data_fungi_mini, q = c(2), first_n = 300)
} # }
if (FALSE) { # \dontrun{
plot_refseq_pq(data_fungi_mini, q = c(2), first_n = 300)
} # }