Graphical representation of distribution of samples across taxa using ridges.
This is the sample-centric counterpart of ridges_pq(): each ridge
represents a taxon (at tax_level) and the x-axis shows the abundance
distribution across samples, optionally colored by a sample factor.
Usage
ridges_sam_pq(
physeq,
fact,
nb_seq = TRUE,
log10trans = TRUE,
tax_level = "Class",
type = "density",
...
)Arguments
- physeq
(required) a
phyloseq-classobject obtained using thephyloseqpackage.- fact
(required) Name of the factor in
physeq@sam_dataused to color the ridges- nb_seq
(logical; default TRUE) If set to FALSE, only the number of samples is counted. Concretely, physeq
otu_tableis transformed in a binaryotu_table(each value different from zero is set to one)- log10trans
(logical, default TRUE) If TRUE, the abundance is log10 transformed.
- tax_level
The taxonomic level used for grouping taxa on the y-axis
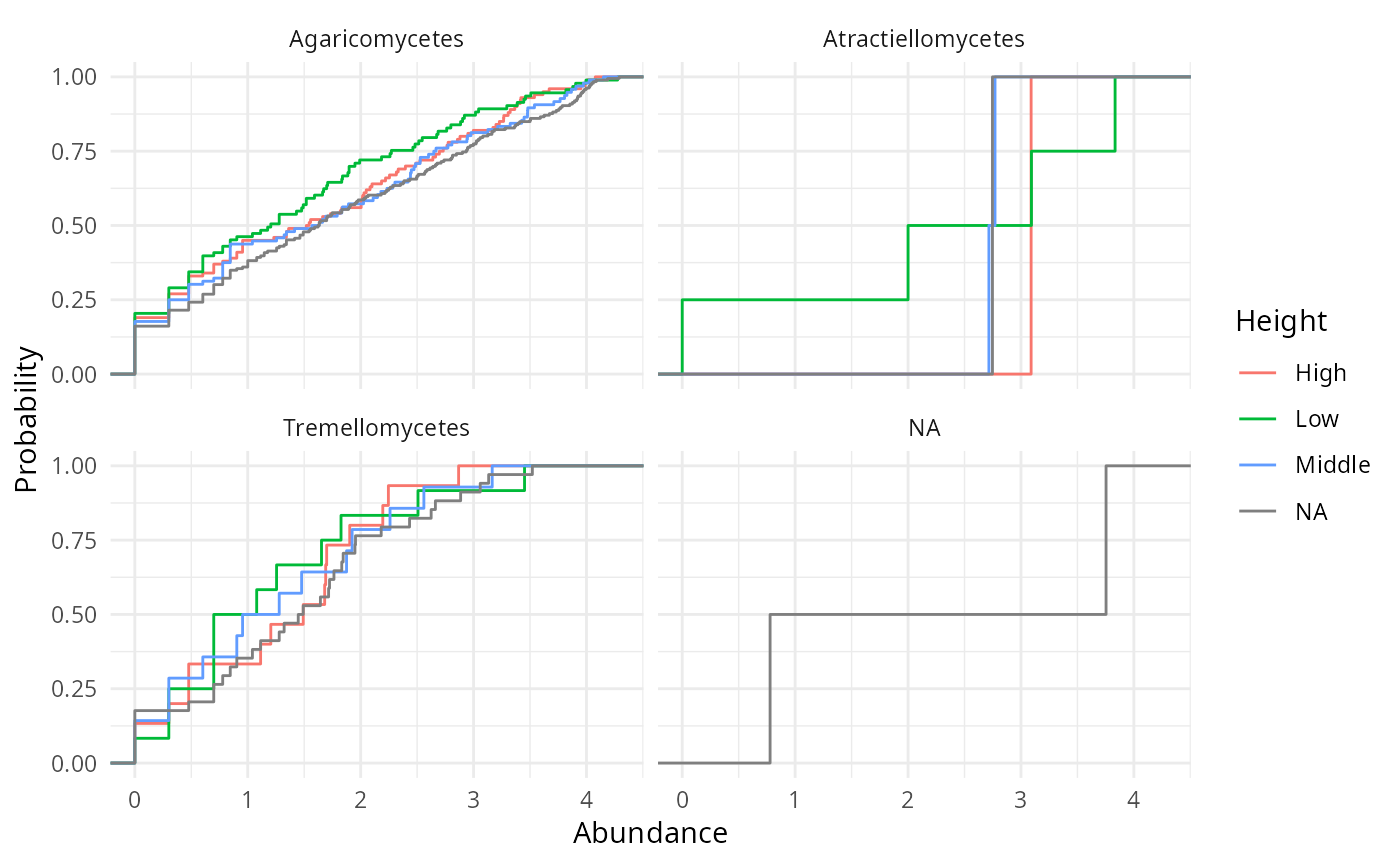
- type
Either "density" (the default) or "ecdf" to plot a cumulative version using
ggplot2::stat_ecdf()- ...
Other params passed on to
ggridges::geom_density_ridges()
Value
A ggplot2 plot with ridges representing the
distribution of samples for each taxon
Examples
if (requireNamespace("ggridges")) {
ridges_sam_pq(data_fungi_mini, "Height",
alpha = 0.5,
log10trans = FALSE, tax_level = "Genus"
) +
xlim(c(0, 1000))
}
#> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Picking joint bandwidth of 36.4
#> Warning: Removed 99 rows containing non-finite outside the scale range
#> (`stat_density_ridges()`).
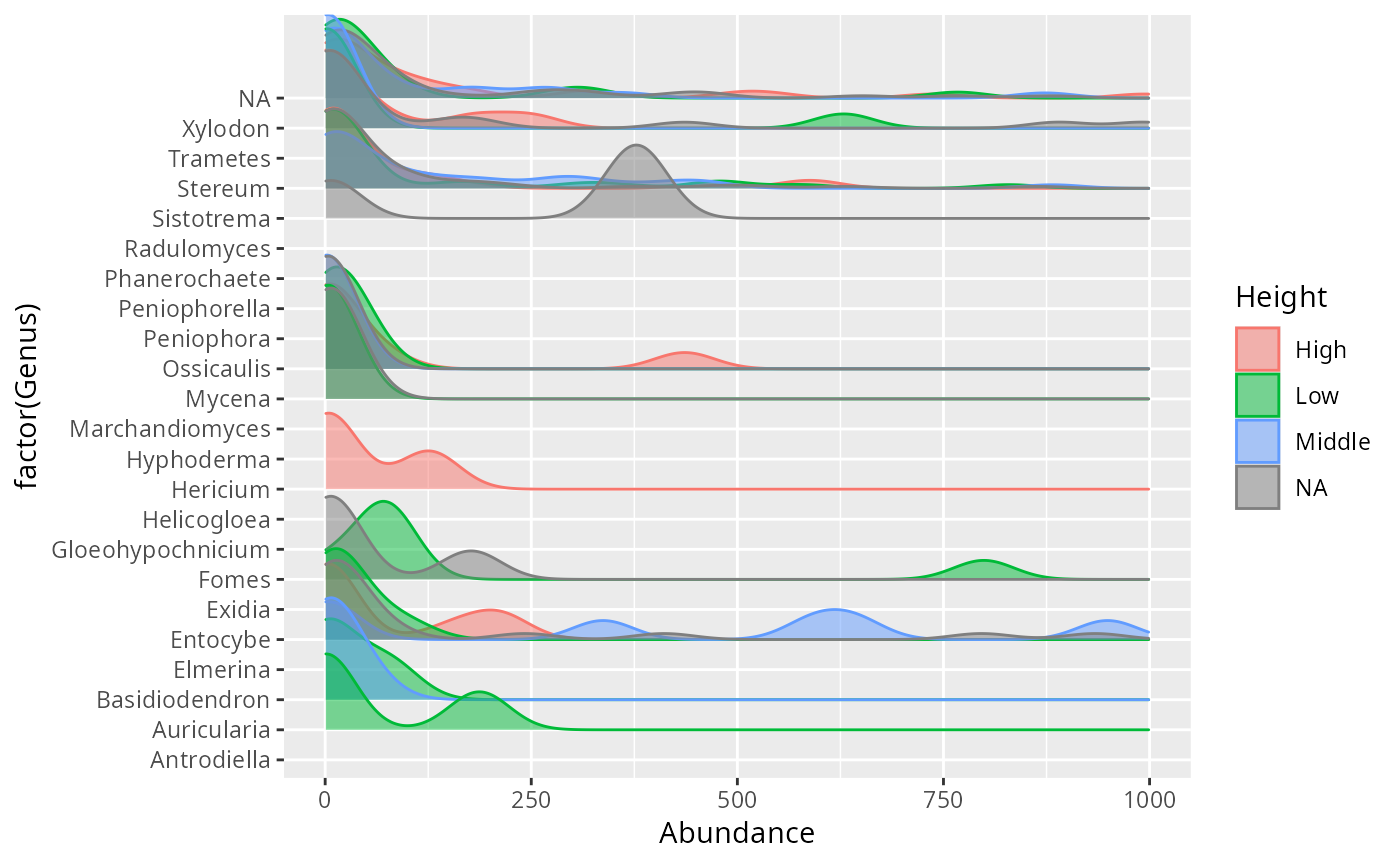 # \donttest{
if (requireNamespace("ggridges")) {
ridges_sam_pq(data_fungi_mini, "Height", alpha = 0.5, scale = 0.9)
ridges_sam_pq(data_fungi_mini, "Height",
alpha = 0.5, scale = 0.9,
type = "ecdf"
)
}
# \donttest{
if (requireNamespace("ggridges")) {
ridges_sam_pq(data_fungi_mini, "Height", alpha = 0.5, scale = 0.9)
ridges_sam_pq(data_fungi_mini, "Height",
alpha = 0.5, scale = 0.9,
type = "ecdf"
)
}
 # }
# }